

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Resistance Mechanisms of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs)
1. Primary resistance mechanism
1) K-Ras mutation
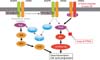
One of the main EGFR downstream signaling molecules is Ras. The activation of EGFR stimulates Ras through the recruitment of a complex consisting of the adaptor proteins Shc and Grb2 and a guanine nucleotide exchange factor for Ras, SOS. Ras activates the downstream Ras/Raf/MEK/ERK pathway, inducing many biological activities involved in tumorigenesis. Mutations of the Ras gene, especially K-Ras, are involved in the pathogenesis and prognosis of lung cancer1. Approximately 10-30% of non-small cell lung carcinoma (NSCLC) patients have K-Ras mutations that are associated with smoking2. The most common mutation of the K-Ras gene is the substitution of the guanine residue in codon 12 to thymine, which causes the constitutive activation of K-Ras. NSCLC patients with K-Ras mutations are associated with unfavorable prognosis.
In 2005, Pao et al.3 reported that K-Ras mutations were associated with a lack of tumor response to EGFR-TKIs. None of the 9 tumors with K-Ras mutations analyzed responded to EGFR-TKI treatment. Furthermore, K-Ras mutations are mutually exclusive with mutations of the EGFR gene and that NSCLC patients with K-Ras mutations have decreased sensitivity to EGFR-TKIs4. An activating Ras mutation could activate the EGFR signaling pathway independent of EGFR activation and might be sufficient to deliver a proliferation or survival signal even if EGFR activity is inhibited by gefitinib treatment (Figure 1)5,6. To test this possibility, two types of gefitinib-sensitive cells (PC9 cells that endogenously express EGFR deletion mutant in exon19 of EGFR gene and HEK293T cells that exogenously express EGFR L858R mutant) were transfected with an expression vector encoding the oncogenic K-Ras G12V mutant. Although the parental cells were sensitive to gefitinib treatment, cells expressing constitutive mutant of K-Ras were less sensitive to gefitinib treatment in a cell growth assay. It was also demonstrated that overexpression of K-Ras induces to activate ERK and/or Akt, promoting S-phase progression and/or suppression of apoptosis, leading to gefitinib resistance. These observations indicate that activated Ras can bypass the inhibition of the upstream EGFR signal and are consistent with the hypothesis that mutational activation of targets immediately downstream of EGFR can induce resistance to gefitinib in lung cancer patients. Thus, it may be beneficial for patients who have K-Ras mutations to avoid EGFR-TKI therapy by screening for K-Ras mutations in cancer tissues.
2) ErbB family members: mutation and amplification
In addition to EGFR, other ErbB family members, including HER2, HER3, and HER4, play important roles during the process of tumorigenesis and in the resistance to EGFR-TKIs because EGFR forms homo or heterodimers with other ErbB family members in response to ligand binding (Figure 1).
Somatic mutations of the HER2 gene were identified in a very small fraction of lung adenocarcinomas7. HER2 mutations are mostly found in females, non-smokers, East Asians, and adenocarcinoma patients. However, the mutations of HER2 are mutually exclusive with those of EGFR in the tumor cells. Most types of HER2 mutations are in-frame insertion mutations in exon 20, leading to constitutively activate the HER2 kinase. It has also reported that HER2 amplification is associated with the sensitivity to EGFR-TKIs in NSCLC patients with EGFR mutations, indicating that HER2 amplification could be associated with gefitinib sensitivity8,9.
HER3 signaling is depends on heterodimerization with other ErbB family members, preferentially HER2 because the tyrosine kinase activity of HER3 is very low. EGFR-mediated activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway requires the activation of PI3K via the dimerization of EGFR with HER3 because HER3 is able to directly couple to PI3K.
Previous reports showed that HER3 mediates the association of EGFR with the PI3K/Akt pathway in gefitinib-sensitive NSCLC cell lines harboring wild-type and mutant EGFRs10 and the expression levels of phospho-HER2 and total HER3 protein are associated with resistance to gefitinib in head and neck squamous cell carcinoma (HNSCC) cell line11.
Gefitinib treatment reduced the phosphorylation of HER3, EGFR, and HER2, consistent with the formation of heterodimers. Furthermore, combination therapy with gefitinib and pertuzumab, an antibody that targets HER2 heterodimerization, provided an additional growth-inhibitory effect compared to gefitinib alone on relatively gefitinib-resistant HNSCC cell lines.
3) Loss of phosphatase and tensin homolog (PTEN)
Activation of Akt occurs by binding to phosphatidylinositol 3,4,5-triphosphate (PIP3), a lipid product of PI3K that acts as a second messenger. Akt activity is also negatively modulated by the PTEN. PTEN dephosphorylates PIP3, reducing PIP3 levels within the cell. Therefore, PTEN acts as a tumor suppressor in opposition to PI3K. Loss of PTEN function provides another potential mechanism of resistance to gefitinib and erlotinib (Figure 1). Somatic mutations of the PTEN gene are often found in various types of human cancers, including NSCLCs. Loss of PTEN function results in Akt hyperactivation caused by an increased concentration of PIP3. Mutations of the PTEN gene in NSCLCs are associated with resistance to EGFR-TKIs12. Bianco et al.13 showed that Akt is activated independent of EGFR signaling in resistant cells, and reintroduction of a PTEN gene restores gefitinib-induced Akt inhibition, indicating that loss of PTEN is associated with acquired resistance. Therefore, PTEN downregulation or Akt activation may be one of the mechanisms for acquired resistance to erlotinib.
2. Acquired resistance mechanism
1) Secondary T790M mutation of the EGFR gene
Unfortunately, many of those patients who originally had responded eventually become insensitive to gefitinib or erlotinib therapy through acquired resistance. The so-called "secondary resistance" usually occurs within a year from the start of the treatment.
A major cause that accounts for approximately 50% of lung adenocarcinoma tissues that acquired resistance cases is the development of a secondary mutation in exon 20 of the EGFR gene. This mutation is a substitution of a hydrophilic threonine residue (T) for a bulkier and hydrophobic methionine (M) in codon 790 (T790M). The presence of a secondary mutation of the EGFR gene in the T790M was reported in 2005. The threonine residue 790 is known as the "gatekeeper residue" and is located in the ATP-binding site, adjacent to the catalytic cleft of the kinase domain14. It was initially proposed that the T790M mutation might prevent the proper binding of tyrosine kinase inhibitors via steric hindrance, similar to the corresponding gatekeeper mutations in BCR-ABL (T315I) and KIT (T670I) that confer resistance to imatinib (Gleevec) in chronic myelogenous leukemia and in patients with gastrointestinal stromal tumors. Interestingly, it was discovered that the T790M mutation does not confer resistance to gefitinib and erlotinib by preventing their binding as originally thought. Instead, the T790M mutation induces resistance by increasing the binding affinity of ATP by one order of magnitude, which is near the affinity for wild-type EGFR, thus enabling ATP to compete effectively with the kinase inhibitors15.
Therefore, the T790M mutation of EGFR is regarded as a marker for acquired resistance to EGFR-TKIs. Tumor cells harboring the EGFR T790M mutation constitute a small minority of the cells before treatment with EGFR-TKIs16. However, after treatment, the T790M mutant tumor cells were found to account for approximately 50% of the cells because of selective growth of T790M mutant cells in response to EGFR-TKIs. Furthermore, human bronchial epithelial cells transfected with an expression vector encoding the T790M EGFR mutant showed a growth advantage over wild-type cells, although not as high as that of deletion mutations in exon 19.
The T790M EGFR mutant exhibited higher level of tyrosine phosphorylation than wild type EGFR, and the T790M/L858R double mutant exhibited a substantial increase in phosphorylation levels compared with the L858R mutant alone17. It has also reported that similar phenomena in both the L858R mutation and a deletion mutation of exon 19 was observed and discussed the importance of the combination of activating mutations and T790M mutations for tumorigenesis18.
In addition, a point mutation in EGFR aspartic acid (D) 761 to Y, L747 to serine (S), and an insertion in exon 20 were reported to exist in lung cancer tissues with acquired resistance to EGFR-TKIs19-21. Balak et al.20 reported that the D761Y mutation modestly reduced the sensitivity of mutant EGFR (L858R) to TKIs. Costa et al.22 also reported that the L747S mutation leads to induce the BH3-only polypeptide Bcl-2-interacting mediator of cell death (also known as BCL2-like 11) and reduced apoptosis induced by EGFR-TKIs. However, this EGFR mutant carrying D761Y or L747S was 100-fold less resistant to EGFR-TKIs than the mutant carrying T790M in vitro.
(1) Irreversible EGFR inhibitors
Second generation inhibitors, designed to address resistance, are currently under investigation in clinical trials23. The two most advanced compounds are dacomitinib (EGFR/HER2/HER4: IC50=6 nM/46 nM/73 nM; Pfizer) and afatinib (EGFR/HER2: IC50=0.5 nM/14 nM; Boehringer-Ingelheim) which are currently in phase III23.
Both of these agents are structurally very similar to gefinitib and erlotinib with the exception that they harbor Michael acceptors in the side chain of the quinazoline core (highlighted in red) (Figure 2)24. This leads to dacomitinib and afatinib to be irreversible inhibitors of EGFR. More specifically, they covalently bind to Cys-797 of EGFR via the sulfur atom (Figure 3). Due to their covalent binding mode, irreversible inhibitors have activity towards EGFR T790M, EGFR T790M/L858R as well as wild-type EGFR. Consequently, dose limitation due to the concomitant inhibition of wild-type EGFR is a potential concern and, in clinical settings, this accompanying inhibition translates into skin rash and diarrhea.
The results of several phase III clinical trials for dacomitinib and afatinib are awaited in 2013. More recently, 'third generation' inhibitors which have an anilinopyrimidine core (such has WZ-4002) (Figures 1, 2) have been identified via high throughput screening of a library of irreversible kinase inhibitors25. Notably, anilinopyrimidine based irreversible inhibitors are 30- to 100-fold more potent against EGFR T790M than other first and second generation inhibitors and are up to 100-fold less potent against wild-type EGFR. Another third generation, mutant-selective inhibitor (CO-1686; Clovis Oncology) entered clinical trials in 2012. Mutant-selective inhibitors, such as WZ-4002 and CO-1686, could potentially offer larger therapeutic windows and better toxicity profiles. It remains to be seen how these third generation inhibitors will behave in the clinic.
2) Amplification of the MET oncogene
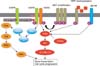
Another mechanism by which patients develop acquired resistance to EGFR inhibition occurs via the amplification of the MET oncogene26,27. In 2007, a cell line with acquired resistance derived from HGC827 (human lung adenocarcinoma) cells that harbor the deletion mutation in EGFR exon 19 was reported. Although the resistant cell line HCG827GR does not have the EGFR T790M mutation, it was found to have MET amplification. MET is a receptor tyrosine kinase that binds to hepatocyte growth factor (HGF) and is associated with the development of various human cancers. In the resistant cells, MET amplification causes the autophosphorylation of MET, its association with HER3, and the activation of the PI3K/Akt pathway (Figure 4)26.
Therefore, EGFR-TKIs fail to inhibit the PI3K/Akt pathway-dependent growth signal because the constitutive phosphorylation of HER3 by MET occurs independent of EGFR activation, which causes the cells to acquire resistance to gefitinib. Thus, upon MET amplification, the activation of the PI3K/Akt pathway leads to cell survival, even under sustained inhibition of EGFR.
MET amplification occurs in approximately 5-20% of the NSCLC patients28. The population with MET amplification was very small before the treatment of EGFR-TKI29,30. However, a small percentage of cells with MET gene amplification selectively grew during the long periods under the EGFR-TKI treatment, and they became a major population, similar to the cells with the T790M mutation.
(1) Met inhibitors (crizotinib, heat shock protein 90 [HSP90])
The concomitant inhibition of MET and EGFR using the monoclonal antiboby MetMab and erlotinib proved to beefficacious in a phase II trial and early clinical trials of small-molecule inhibitors of MET given with EGFR inhibitors are now currently underway (Figure 5).
The combination of crizotinib (dual c-MET/anaplastic lymphoma kinase [ALK] inhibitor) (Figure 5) and dacomitinib, although in early phase, is particularly interesting since one of its components, dacomitinib, was designed to address the acquired resistance by the T790M secondary mutation in EGFR. This combination could potentially be effective in patients with acquired resistance via T790M and/or MET amplification.
Another potential strategy to overcome acquired resistance is the use of combination therapies including HSP90 inhibitors. It has been shown that several EGFR mutants, such as EGFR L858R/T790M, are client proteins of molecular chaperone HSP90. Additionally, MET is also a client protein of HSP90. Two structurally diverse classes of HSP90 inhibitors have been looked at in clinical settings: the ansamycin-like derivatives such as IPI-50429 and bi-phenolic derivatives such as AUY92230 and STA9090.31. A combination of AUY922 and erlotinib is currently in clinical trials in patients with acquired resistance (Figure 5).
3) Overexpression of HGF
Not only MET amplification but also the overexpression of HGF has been reported as a mechanism of EGFR-TKI resistance in 200831. In cancer tissues from lung adenocarcinoma patients who showed resistance to gefitinib, high expression levels of HGF were observed in the lung cancer cells that did not harbor the T790M mutation or MET amplification. Overexpression of HGF was shown to induce resistance to gefitinib or erlotinib in lung adenocarcinoma cells harboring the EGFR-TKI-sensitive mutation. Unlike MET amplification, overexpression of HGF stimulates the PI3K/Akt pathway through MET phosphorylation independent of ErbB3 phosphorylation (Figure 4).
In 2009, Wang et al.32 showed that gefitinib-sensitive lung cancer cells became resistant to gefitinib when co-cultured with HGF-producing fibroblasts. Furthermore, the resistance induced by fibroblast-derived HGF was abolished by anti-HGF antibodies or HGF antagonists, such as NK432. It was suggested that such HGF inhibitors could be used to overcome the resistance to EGFR-TKIs.
The analysis of 16 clinical specimens from NSCLC patients with acquired resistance revealed that MET amplification was present in 25% (4 of 16) of tumor specimens after gefitinib treatment, although an extremely low level (<1%) of MET amplification was detected in these 4 cases before gefitinib treatment. Moreover, in all the 4 cases with MET amplification, a higher expression of HGF was observed after gefitinib treatment than before treatment. These results indicate that HGF accelerates the expansion of the tumor cells with MET amplification. The cells with MET amplification as the T790M mutation was present in a small proportion of the tumor cells before EGFR-TKI treatment and became dominant during treatment with EGFR-TKIs, suggesting that HGF may play an important role in this process.
Onitsuka et al.33 examined clinical specimens of the acquired resistance cases and found that certain lung adenocarcinoma tissues have the T790M mutation and high levels of HGF expression. Turke et al.34 reported similar results. These results indicate the coexistence of the T790M mutation and HGF overexpression in patients with acquired resistance to EGFR-TKIs.
4) Insulin-like growth factor 1 (IGF-1) receptor signaling
Sharma et al.35 suggested chromatin modification as a novel mechanism of acquired resistance to EGFR-TKIs. In their study, these authors established gefitinib-resistant cell lines derived from lung adenocarcinoma PC9 cells that harbor a gefitinib-sensitive exon 19 deletion. The treatment of the PC9 cells with gefitinib induced extensive cell death within 9 days, but the cells expressing the stem cell associated surface antigen CD133 were spared. Although most of these cells were quiescent, approximately 20% of them resumed normal growth in the presence of gefitinib.
After these cells were cultured in drug-free medium, they reacquire the sensitivity to EGFR-TKIs, indicating that the resistance of these cells is reversible. The mechanism of reversible resistance was concluded to be associated with the enhancement of IGF-1 receptor signaling. In the resistant cells, the histone demethylase RBP2/KDM5A/Jarid1A is upregulated, and causes activation of insulin-like growth factor 1 receptor (IGF1R). In addition to erlotinib, treatment with IGF1R inhibitors restores sensitivity to EGFR-TKIs. Guix et al.36 detected the hyperphosphorylation of IGF1R in gefitinib-resistant cells derived from squamous cell lung carcinoma. These reports indicate that IGF1R signaling may be involved in the resistance to EGFR-TKIs in some cases (Figure 4).
5) PIK3CA mutations and Akt activation
The PI3K protein family comprises lipid kinases that can regenerate phosphatidylinositol-3-phosphate, which is a key mediator between growth factor receptors and intracellular downstream signaling pathways including the EGFR family of receptors (Figure 1)37. Mutation in the main catalytic subunit of the PI3K (PIK3CA) has been observed as an acquired resistance mechanism to EGFR-TKIs in vitro38. Sequist et al.28 detected that 2 out of 37 EGFR mutation-positive patients developed PIK3CA mutations after progression while on EGFR-TKI therapy (Figure 6). Other studies have shown that PIK3CA mutations occurred in about 4% of NSCLCs39 and have been observed in both squamous and adenocarcinoma40. PIK3CA mutations are not mutually exclusive of EGFR or KRAS mutations. Interestingly, PIK3CA mutations have been observed in patients with EGFR mutations without prior exposure to EGFR-TKIs. In one study, all 4 patients with PIK3CA mutations had coexisting EGFR mutations, and these mutations were found in exons 9 and 2041. Akt activation is found in 30-75% of NSCLC and in 2% of cases (limited to squamous cell subtype) an E17K point mutation of Akt1 leads to its PI3K-independent activation. Akt activation is a poor prognostic factor and has implicated in resistance to chemotherapy and radiation.
6) Small-cell lung cancer (SCLC) transformation
The same Sequist et al.28 study discovered five EGFR mutation positive adenocarcinoma patients "transformed" to SCLC after progression on treatment with first generation EGFR-TKIs (Figure 6). These "transformed" SCLC patients retained the original EGFR mutation and most also responded to SCLC-based platinum-etoposide therapy. This seminal finding indicated that resistance to EGFR-TKIs in EGFR mutation-positive patients is dynamic, and repeat biopsy on progression may help guide subsequent treatment.
7) Epithelial-to-mesenchymal transition (EMT)
Three out of the seven patients from the same Sequist et al.'s study28 who did not develop any identifiable mutations showed EMT at the time of TKI resistance (Figure 6).
When compared with pre-treatment samples, two of the three patients' tumor samples showed acquired vimentin expression and loss of E-cadherin expression. EMT has been shown in vitro to confer resistance to EGFR-TKIs in NSCLC cell lines42.
2. Resistance mechanism for ALK inhibitors
This fusion gene encodes for the cytoplasmic fusion protein EML4-ALK which, upon dimerization, results in constitutive activation of the kinase domain of ALK. The downstream pathways activated by ALK include PI3K/Akt, MEK/ERK and the JAK/STAT pathways which lead to cell proliferation and cell survival43. It is estimated that approximately 5% of all NSCLC cases have EML4-ALK fusions. Although this appears to be a relatively small percentage, because of the high prevalence of lung cancer cases, it would represent about 70,000 patients worldwide and close to 10,000 in the USA alone44.
Similar to the NSCLC patients harboring activating EGFR mutations, ALK-positive patients are generally non smokers with adenocarcinoma histology45. Strikingly, only 4 years following the discovery of the EML4-ALK fusion gene, and the subsequent in vitro and in vivo pharmacological target validation confirming the oncogenic activity of its encoded protein46, EML4-ALK-positive patients were already taking advantage of ALK inhibitors. For instance, in phase I/II clinical trials, 250 mg twice-daily oral administration of crizotinib, a dual c-MET/ALK inhibitor, to patients with advanced ALK-positive NSCLC led to an overall response rate of 57% and the progression-free survival at 6 months was estimated to be 72%47. Notably, all patient tumors tested were negative for MET amplification, suggesting that clinical responses seen with crizotinib is through ALK inhibition. In August 2011, following these positive results, the Food and Drug Administration (FDA) approved crizotinib (Xalkori, Pfizer) for patients with advanced ALK-positive NSCLC as diagnosed using fluorescence in situ hybridization (FISH) with the FDA-approved Vysis FISH probe kit (Abbott).
1) Acquired resistance of ALK inhibitors
Crizotinib, an ALK-TKI, responds well to EML4-ALK lung cancer (60% or more) and has therapeutic efficacy comparable to that of EGFR-TKI in treating EGFR-mutant lung cancer. However, almost every patient who responds well to crizotinib will, without exception, experience recurrence due to acquired resistance48.
The mechanisms for acquired resistance include gatekeeper L1196M mutations49, ALK gene amplification50, and other ALK gene mutations (L1152R, C1156Y, F1174L)51-53.
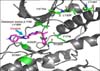
The first two secondary ALK mutations that conferred resistance to crizotinib were reported within the same issue of the New England Journal of Medicine describing the positive clinical results mentioned above. The molecular analysis of a patient's tumor, who relapsed only 5 months after the beginning of its treatment, revealed two distinct mutations. Specifically, a leucine to methionine mutation at position 1196 (L1196M) and a cysteine to tyrosine mutation at position 1156 (C1156Y) were found. The L1196M mutation corresponds to the 'gatekeeper' mutation (analogous to the T790M mutation in EGFR), whereas the C1156Y mutation is located in the a-C-helix near the upper edge of the ATP-binding site (Figure 7)49.
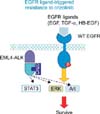
With regard to resistance due to ligands, EGFR ligands (epidermal growth factor, amphiregulin, HB-EGF, and transforming growth factor-α) are reported to induce crizotinib resistance. Sasaki et al.52 reported that cells derived from pleural effusions of patients with EML4-ALK lung cancer who developed acquired resistance to crizotinib displayed the gatekeeper L1196M mutation and high levels of expression of EGFR ligands (EGF and amphiregulin). This suggests the existence of a mechanism of resistance due to activation of an EGFR pathway in an autocrine manner. Exogenously adding EGFR ligands activated EGFR pathway and induced resistance to crizotinib in a paracrine manner (Figure 8)54,55.
The two mutations were shown to independently induce crizotinib resistance in Ba/F3 cells engineered to express the L1196M and C1156Y mutations. By mid-2012, additional patient studies led to the discovery of several other secondary mutations56,57. That is, three other mutations in the a-Chelix were discovered: a threonine insertion at amino acid 1151 (1151Tins), a leucine to arginine (L1152R) and phenylalanine to leucine (F1174L). Two other mutations have been discovered at the solvent interface: a glycine to arginine (G1202R) and a serine to tyrosine (S1206Y). And finally, a glycine to alanine mutation (G1269A) was found in the ATP binding pocket (Figure 7). The resistance to ALK inhibitors appears, for now, much more complex since a greater variety of mutations with similar degrees of frequency is found in patients. This is reminiscent of BCR-ABL, where almost 20 different secondary mutations within the kinase domain have been found58.
Conclusion
The past 5 years have witnessed tremendous discoveries of specific driver mutations in NSCLC, and specific inhibitors to the driver mutations are being investigated to target these subsets of patients, with EGFR TKIs and ALK inhibitors (crizotinib). Also, mechanisms for resistance to molecularly targeted drugs have gradually been clarified, and strategies to treat that resistance have been indicated. Therefore, optimal treatment for each patient requires accurate diagnosis of the mechanism by which the individual patient developed resistance. In such instances, a second biopsy is recommended, but in the event of multiple lesions all of their origins would have to be biopsied, a feat that would be rather difficult. A highly reliable, minimally invasive diagnostic technique needs to be developed to achieve that goal.




 XML Download
XML Download