

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Diffuse alveolar hemorrhage (DAH) is a life-threatening condition caused by a variety of disorders associated with hemoptysis, anemia, diffuse lung infiltration, and acute respiratory failure. DAH originates from the pulmonary microcirculation, including the alveolar capillaries, arterioles, and venules and is usually diffuse, but may also be focal. DAH should be distinguished from other causes of pulmonary hemorrhage caused by localized pulmonary abnormalities and the bronchial circulation. Early bronchoscopy with bronchoalveolar lavage (BAL) is generally required to confirm the diagnosis of DAH and rule out infection.
Systemic vasculitis is one of the most common causes of DAH and can be pathologically defined by the presence of cellular inflammation, vessel destruction, tissue necrosis, and eventually, organ dysfunction. The lung is the site frequently involved in systemic vasculitis. The clinical features of each patient's disease are determined by the site, size, and type of vessel involved.
Because the clinical presentation of the disease underlying DAH is highly variable, the identification of a cause is very difficult. Thus, particular attention should be paid to the pattern of findings or clinical scenarios. The diagnosis of DAH relies on the clinician's recognition combined with clinical, laboratory, radiologic, and pathologic features.
Early recognition is crucial because prompt diagnosis and treatment are required for survival. Despite advances in the identification and management of DAH, it remains a condition with high morbidity and mortality. This article aims to provide a general review of the clinical presentation and causes of DAH and to recommend a diagnostic approach and management plan for the most common causes.
Definition
DAH is a distinct clinicopathologic syndrome of pulmonary hemorrhage that originates from the pulmonary microcirculation, including the alveolar capillaries, arterioles, and venules. It presents with hemoptysis, anemia, diffuse lung infiltration, and acute respiratory failure. The diagnosis is confirmed by the observation of the accumulation of red blood cells (RBCs), fibrin, or hemosiderin-laden macrophage in the alveolar space on pathologic biopsy1. Hemosiderin, a product of hemoglobin degradation, appears at least 48-72 hours after bleeding and is helpful in distinguishing DAH from surgical trauma. Mild interstitial thickening, organizing pneumonia, or diffuse alveolar damage can also be seen2.
DAH is associated with pulmonary capillaritis, bland pulmonary hemorrhage, diffuse alveolar damage, and miscellaneous histology. Pulmonary capillaritis is the most common finding3. Pulmonary capillaritis has a unique histopathologic appearance, consisting of an interstitial neutrophilic predominant infiltration, fibrinoid necrosis of the alveolar and capillary walls, leukocytoclasis caused by systemic vasculitis, anti-glomerular basement membrane (GBM) disease, and classic autoimmune disease2. The infiltrating neutrophils undergo cytoclasis and nuclear debris accumulates within the interstitium, and there is a subsequent loss of the integrity of the alveolar capillary basement membrane. Systemic vasculitis can also involve the microcirculation. Pulmonary capillaritis may also be a small vessel vasculitis limited to the lung. Pulmonary vasculitis refers to inflammation of the lung vessels of any size, whereas pulmonary capillaritis is confined to the microcirculation of the lung3.
Etiology and Classification
A number of diseases can cause DAH. In general, DAH occurs in three characteristic patterns which reflect the nature of the underlying vascular injury (Table 1). Any source of injury to the alveolar microcirculation can theoretically cause alveolar hemorrhage4,5.
1. DAH associated with vasculitis or capillaritis
Most cases of DAH are caused by pulmonary capillaritis and are closely associated with systemic vasculitis and findings such as anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, anti-GBM disease, systemic lupus erythematosus (SLE), and collagen vascular diseases (CVDs). It is also seen with several other conditions, including the use of certain drugs and transplantation.
2. Bland pulmonary hemorrhage (without capillaritis or vasculitis)
In this pattern, RBCs leak into the alveoli without any evidence of inflammation or destruction of the alveolar capillaries, venules, and arterioles. The epithelial lesions are usually microscopic and are scattered geographically. Anti-GBM diseases and SLE can induce both pulmonary capillaritis and bland pulmonary hemorrhage.
Clinical Presentations, Symptoms, and Signs
DAH can be defined by the presence of hemoptysis, diffuse alveolar infiltrates, a drop in hematocrit, and hypoxemic respiratory failure. Hemoptysis may develop for a few hours or a few days, but up to one-third of patients do not have hemoptysis. The alveolar infiltrates can be unilateral, and a drop in hematocrit or hemoglobin can be difficult to document. DAH may be presented in acute, subacute, or repetitive patterns with variable severity. Therefore, DAH must be considered in patients with otherwise unexplained alveolar infiltrates. Non-specific cough, dyspnea, chest pain, and fever may develop. Underlying systemic symptoms may also be present. Thus, a high index of clinical suspicion is essential. Two possible clinical scenarios are described in the following sections4.
1. DAH with associated systemic symptoms and signs
Certain clues from a patient's clinical history that should raise suspicion for DAH are: 1) recent infection suggesting Henoch-Schönlein purpura or cryoglobulinemic vasculitis, 2) use of a possibly offending drug such as an anticoagulant, D-penicillamine, nitrofurantoin, amiodarone, propylthiouracil, cocaine, or sirolimus, 3) exposure to toxic agents such as trimellitic anhydride, insecticides, or pesticides, or 4) a known co-morbid condition such as systemic vasculitis, CVD, mitral valve disease, or solid organ or stem cell transplantation4.
Clinical scenarios suggestive of vasculitis are: 1) DAH, 2) acute glomerulonephritis (GN), 3) pulmonary-renal syndrome, 4) deforming or ulcerating upper airway disease, 5) cavitary or nodular disease on chest imaging, 6) palpable purpura, 7) mononeuritis multiplex, or 8) multisystem disease6.
If sinus disease, skin manifestations, pulmonary parenchymal nodules, and cavitary lesions coexist with positivity for anti-proteinase 3 (PR3) C-ANCA and biopsy-proven granuloma, then Wegener's granulomatosis (WG) should be considered. DAH with GN and skin manifestations, positivity for P-ANCA, and necrotizing non-granulomatous lesions on end-organ biopsy may lead to a diagnosis of microscopic polyangiitis (MPA). Churg-Strauss syndrome (CSS) should be considered if asthma, eosinophilia, pulmonary infiltrates, and DAH coexist. In young smokers with GN and DAH presenting as either bland alveolar hemorrhage or pulmonary capillaritis, Goodpasture's syndrome or anti-GBM disease are possible diagnoses.
2. DAH without systemic symptoms and signs
When the above conditions have been considered but no suggestive findings are found, the following four conditions should be considered: 1) anti-GBM disease in limited pulmonary form or onset, for which positivity to the antibody with linear deposits in the lungs would be diagnostic, 2) pulmonary-limited MPA, which would be positive for positive anti-myeloperoxidase (MPO) P-ANCA, 3) pauci-immune isolated pulmonary capillaritis, which would show evidence of neutrophilic pulmonary capillaritis upon biopsy, or 4) idiopathic pulmonary hemosiderosis, a diagnosis of exclusion, when the biopsy shows evidence of acute, subacute, and chronic bland DAH and no evidence of vasculitis in young age4.
Diagnostic Approach
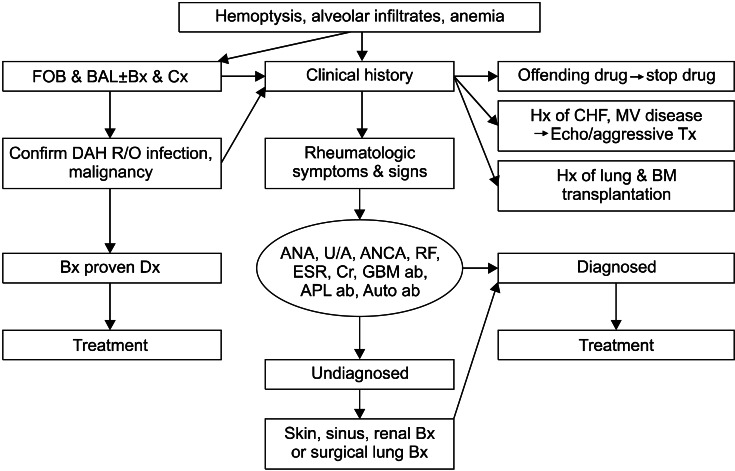
Since DAH represents a medical emergency, a thoughtful and thorough approach to the diagnosis of DAH is critical to appropriate management. The diagnosis of DAH relies on the clinician's recognition combined with specific clinical, laboratory, radiologic, and pathologic features. The two important goals of the clinical evaluation are: 1) establishing the diagnosis of DAH and 2) identifying the underlying cause7. Although individual cases of DAH may require modification, the recommended approach is outlined in the following sections (Figure 1).
1. Careful history taking and physical examinations
Careful attention should be given to the presence of symptoms such as hemoptysis, cough and dyspnea, and any pre-existing medical conditions. A detailed drug and occupational history should be taken. In patients who present with hemoptysis, focal sources of pulmonary hemorrhage and upper airway and gastrointestinal bleeding sources must be excluded. Congestive heart failure, pneumonia, and other acute presentations of diffuse parenchymal lung disease must be also considered1.
A careful evaluation of the eyes for evidence of episcleritis or retinal vasculitis, the nasopharynx for nasal septal erosion or saddle nose deformities, and the skin for leukocytoclastic vasculitis should be conducted. DAH may represent the primary manifestation of an underlying systemic vasculitis or CVD, so the absence of historical or physical evidence should not eliminate these as possible causes1.
2. Laboratory studies
In initial diagnostic studies, culture of blood and other affected organs must be obtained to exclude infection. Although the findings are generally nonspecific, routine laboratory tests such as a complete blood count with differential, chemistry, liver tests, blood urea nitrogen, and creatinine should be obtained. An elevated erythrocyte sedimentation rate and C-reactive protein are expected in active vasculitis but lack specificity. Urinalysis with microscopic examination should be obtained in all patients. Proteinuria and microscopic hematuria are common early findings in WG and MPA. Anti-GBM disease should be screened in all patients with DAH or a pulmonary-renal syndrome. In most patients with DAH, serum ANCA, anti-GBM antibodies, antinuclear antibody (ANA), and anti-phospholipid antibodies should be obtained.
ANA and rheumatoid factor may be positive in primary vasculitis, but high titers and disease-specific antibodies such as dsDNA, SS-A/SS-B, anti-ribonucleoprotein, and anti-JO-1 favor a CVD. Total IgE should be obtained when CSS is being considered. Complement (C3 and C4) should be obtained whenever SLE is in the differential6.
C-ANCA is primarily associated with antibodies directed against PR3. The P-ANCA pattern is observed in the context of antibodies directed against a wide variety of intracellular antigens; it is most commonly associated with MPO. C-ANCA is highly sensitive (90-95%) in active, systemic WG, with a specificity of approximately 90%. However, a positive P-ANCA lacks sensitivity and provides no more than suggestive evidence of CSS, MPA, or idiopathic pauci-immune GN because it can be found in a wide variety of settings, including rheumatoid arthritis and Goodpasture's syndrome6.
In stable patients, the diffusion capacity for carbon monoxide can be measured and, if elevated, favors a diagnosis of DAH8. Recent alveolar hemorrhage increases the diffusion capacity for carbon monoxide, whereas hemorrhage that occurs more than 48 hours before testing is unlikely to cause significant elevation.
3. Radiologic findings
Plain chest X-rays and computed tomography scans often show abnormalities even in the absence of clinically significant symptoms. Chest X-rays may show patchy or diffuse alveolar opacities. Recurrent episodes of hemorrhage may lead to reticular interstitial opacities due to pulmonary fibrosis, usually with minimal honeycombing. Computed tomography may show areas of consolidation interspersed with areas of ground-glass attenuation and preserved normal areas. These findings are generally nonspecific. Cavities, nodules, and diffuse ground glass opacification with DAH is suspicious of vasculitis. Lymph node adenopathy is not a common finding and is more suggestive of infection or malignancy4,6,9.
4. Bronchoscopy
Early bronchoscopy is indicated in most patients who are suspected to have DAH. Bronchoscopy is used primarily to document alveolar hemorrhage and exclude infection. A rising RBC count in sequential BAL aliquots from the same location is considered diagnostic of DAH. BAL specimens should be sent for routine bacterial cultures and fungal, viral, and Pneumocystis carinii studies when indicated. The use of transbronchial biopsy (TBB) in patients with suspected DAH is controversial2. Due to the small size of the specimens and the mechanical disruption of tissue architecture that generally occurs, TBB is rarely used in identifying the cause of DAH10.
5. Diagnostic biopsy
Although a confident diagnosis may occasionally be made without tissue biopsy, diagnostic biopsy remains key to diagnosis. Diagnostic tissue may be obtained from easily accessible sites, such as the skin or upper airway lesions10.
If systemic vasculitis, Goodpasture's syndrome, or CVD is suspected, renal biopsy is preferred and should be obtained immediately. In addition to conventional histopathology, immunofluorescence (IF) and electron microscopy studies should be performed when appropriate. A renal biopsy with direct IF is usually helpful if there are laboratory abnormalities suggestive of renal insufficiency or GN. Light microscopy showing focal segmental necrotizing GN is not by itself sufficient to differentiate between the GN seen in WG, MPA, Goodpasture's syndrome, or SLE. However, direct IF will show clumpy immune complex deposition in SLE, linear immune complex deposition along the basement membranes in Goodpasture's syndrome, and scanty or no immune deposits in the pauci-immune GN typical of MPA or WG2.
In patients in whom the diagnosis of DAH is still in question after a thorough clinical evaluation or in whom the underlying cause of DAH is still unclear after appropriate serologic testing, surgical biopsy should be entertained. When the lung is clinically involved, surgical lung biopsy almost always provides definitive pathologic evidence. Although useful in confirming DAH, surgical lung biopsy is generally unable to identify the underlying cause11. The biopsy samples should be processed so as to have tissues in saline for culture, frozen tissue for IF, and formalin-fixed tissue for standard hematoxylin and eosin staining.
Treatment and Management
Therapy for DAH consists of treating both the autoimmune destruction of the alveolar capillary membrane and the underlying condition. Corticosteroids (CS) and immunosuppressive agents remain the gold standard for most patients. Recombinant-activated human factor VII seems to be a promising new therapy, but further evaluation is needed.
Immunosuppressive agents are the mainstay of therapy for DAH, especially if associated with systemic or pulmonary vasculitis, Goodpasture's syndrome, or CVDs. Most experts recommend intravenous methylprednisolone (Solu-Medrol) at up to 500 mg every 6 hours, although lower doses seem to have similar efficacy, for 4 or 5 days, followed by a gradual taper to maintenance doses of oral steroids4.
Other immunosuppressive drugs such as cyclophosphamide (CYC), azathioprine (AZA), methotrexate (MTX), mycophenolate mofetil (MMF), and etanercept may be used in DAH, especially in severe cases refractory to first-line therapy with CS. Plasmapheresis (PE) is indicated for DAH associated with Goodpasture's syndrome or other vasculitic processes in which the titers of pathogenic immunoglobulins and immune complexes are very high.
Before the institution of immunosuppressive therapy, patients with systemic vasculitis had a mortality rate of 75%. CS therapy alone improved mortality, but the 5-year mortality remained at 50%. A major breakthrough occurred when CYC was added to CS, which lowered the 5-year mortality to 12%12. Despite this impressive progress, the mortality of patients with systemic vasculitis who receive treatment still remains high. Therefore, the goals of therapy in patients with systemic vasculitis are the prevention of disease-related mortality or morbidity and the minimization of treatment-related complications.
1. General principles
The backbone of therapy for vasculitis is the early identification of disease followed by the rapid initiation of disease control with immunosuppression. The severity of the disease generally dictates the intensity of the initial treatment and the risk of complication. Therapy is often divided into two phases: An initial "remission-induction" phase controls active disease, and a "maintenance" phase, which uses less intensive therapy, maintains disease remission while lowering the risk of adverse medication related events.
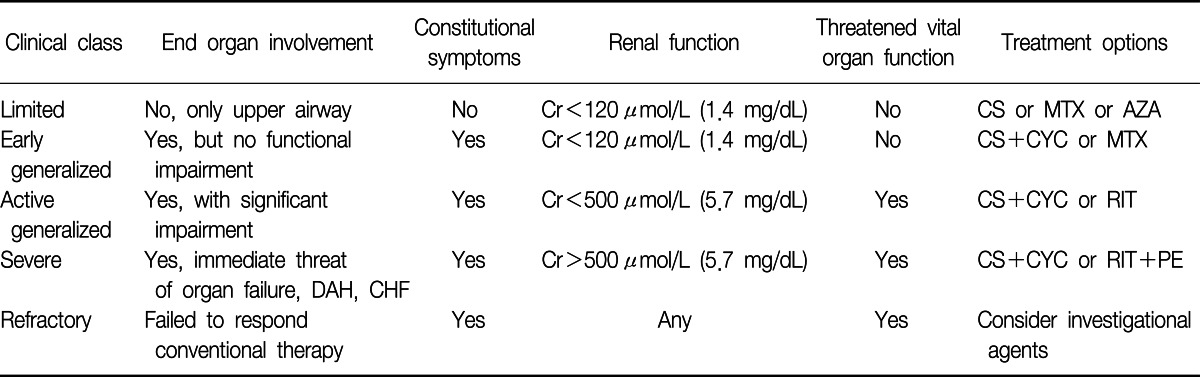
Treatment recommendations depend on an accurate determination of disease severity. Although severity and prognosis are dependent on a number of factors, the most important of which seems to be the severity of disease activity as measured by the number of organ systems involved, the degree of renal disease, and the presence of DAH. Based on these associations, the European Vasculitis Study Group (EUVAS) has devised a clinically useful grading system (Table 2) in which the patient's disease is categorized as 1) limited, 2) early, generalized, 3) active, generalized, 4) severe, or 5) refractory13,14.
2. Remission-induction therapy
1) Limited disease
Limited disease refers to localized disease of the upper airway. These patients have no systemic symptoms, no impairment of end organ function, and no renal involvement. In this setting, therapy can often be limited to a single agent, such as CS, AZA, or MTX.
2) Early, generalized disease
Early, generalized disease is differentiated from active generalized disease by the presence or absence of impaired organ function. Treatment recommendations for these two subsets have traditionally been similar, with CYC plus CS being the first-line therapy for both conditions. MTX is also an acceptable first-line therapy for early generalized disease, and, given the potentially more favorable side-effect profile, the combination of MTX plus CS is increasingly being used.
3) Active, generalized disease
CYC plus CS has remained the principal first-line therapy for the treatment of active, generalized vasculitis12. Recent evidence suggests that pulsed intravenous CYC may be as effective as oral CYC while having fewer side effects.
4) Severe disease
Severe disease is defined by the functional impairment of critical organs, such as severe renal disease (creatinine>5.7 mg/dL), DAH, or other life-threatening disease. Recent studies suggest that patients with severe disease should receive a combination of CYC, CS, and PE therapy. The addition of PE therapy to the standard CYC plus CS regimen has been shown to be superior to high-dose, pulsed, intravenous steroids at restoring renal function in patients with severe renal impairment and DAH15. Additional therapy, described in a case report, for patients with intractable DAH include activated human factor VII, which was used to induce hemostasis16. As described in a case report, extracorporeal membrane oxygenation has also been used to buy time until the onset of action of other interventions17.
5) Refractory disease
Patients who have not responded to the aggressive use of cytotoxic agents, high-dose CS, or PE are deemed to have refractory disease. For this group, the physician must consider the use of novel or experimental agents. Treatments such as intravenous immunoglobulin, anti-tumor necrosis factor-α therapy with infliximab, T-cell depletion therapy with antithymocyte globulin, and B-cell depletion therapy with rituximab (a monoclonal anti-CD20 antibody) have been used. In a recent series of 11 patients with active ANCA-associated vasculitis, rituximab was used to treat patients who had received maximal doses or had other contraindications to CYC therapy. Remission was induced in all 11 patients, and ANCA titers became undetectable in eight patients18.
3. Maintenance therapy
Maintenance therapy used to maintain control of the disease requires less immunosuppression and should be associated with fewer and less severe adverse effects. After the induction of clinical remission with an agent such as CYC, patients generally convert to AZA or MTX19,20. Additional agents that have been used in select patients include MMF, leflunomide, and cyclosporine. However, etanercept was not effective for the maintenance of disease remission in WG patients21. In the absence of a disease flare, maintenance therapy is generally continued for 12 to 18 months. A longer duration of therapy should be considered for patients at high risk for relapse.
Specific Disorders and Causes of DAH
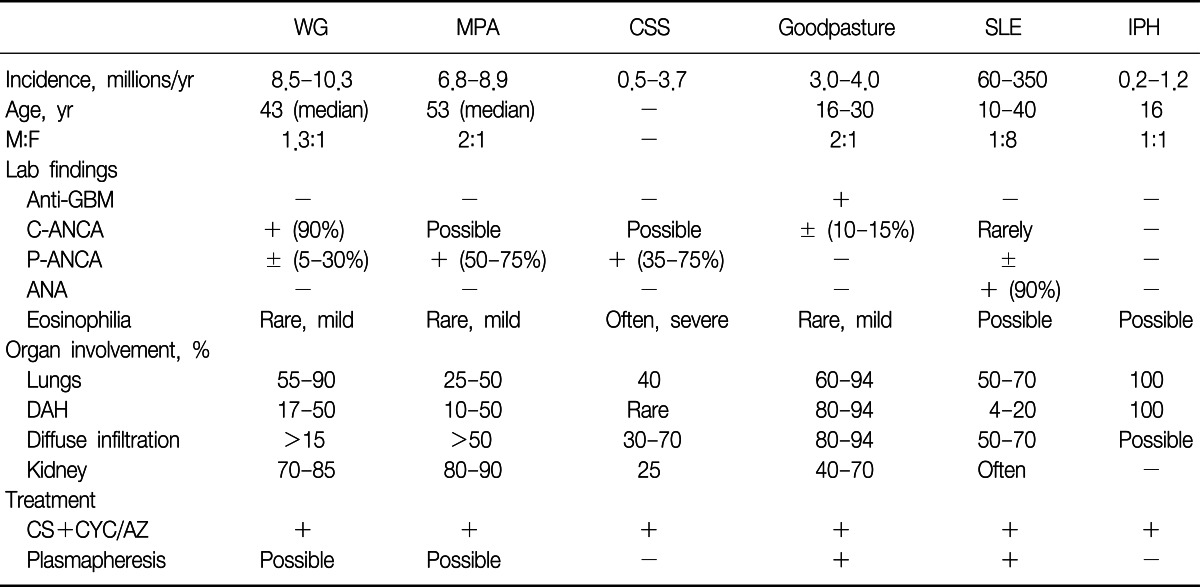
Table 3 summarizes the common causes and diagnostic features of DAH.
1. Wegener's granulomatosis
WG is a systemic vasculitis that primarily affects middle-aged adults and is characterized by necrotizing granulomatous inflammation. In 1936, Wegener published a report on a new disease that would be differentiated from periarteritis nodosa. In 1954, Godman and Churg described a detailed diagnostic triad classic for Wegener's consisting of necrotizing granulomatous inflammation of the upper and lower respiratory tract, generalized necrotizing vasculitis involving the small arteries and veins, and necrotizing GN22,23.
Of the ANCA-associated vasculitis, WG is the most common. The upper and lower respiratory tracts are typically affected, with destruction of the nasal septum and sinuses and cavitating lesions in the lungs. Involvement of the kidney is common, with histopathologic lesions of focal segmental necrotizing GN. The eyes, skin, and other organs may also be involved. Frequently, the complete triad is not present at initial presentation. The diagnosis of WG is commonly made serologically, with a positive C-ANCA. A positive C-ANCA or anti-PR3 is found in 85% to 95% of active systemic disease but is less frequently seen (60-65% sensitive) in organ-limited disease and is even less common (40% sensitive) in remissions. Its specificity is approximately 90%6.
In cases in which the diagnosis remains uncertain, surgical biopsy of the affected organs (primarily the lungs) may show typical necrotizing granulomatous lesions. DAH either can complicate an established case of WG or represent the initial manifestation of the disease. DAH is often subclinical and recurrent in WG. This pattern of frequently recurring DAH is seen with ANCA-associated vasculitis and CVD1.
Treatment with high-dose CS and CYC is the initial therapy recommended for DAH that complicates WG. AZA may be substituted for CYC after remission. Intravenous immunoglobulin may be effective for persistent disease. Mortality is considerable and most often caused by acute respiratory failure, renal failure, or superimposed infection as a result of immunosuppressive therapy. Poor outcomes are correlated with advanced age, more severe renal impairment, alveolar hemorrhage, and anti-PR3 positivity.
2. Allergic Angitis with Granuloma (CSS)
In 1951, Churg and Strauss reported a new entity separate from periarteritis nodosa presenting with fever, severe asthma, and hypereosinophilia, with evidence of systemic vasculitis. The syndrome is characterized by the triad of 1) asthma, 2) hypereosinophilia, and 3) necrotizing vasculitis. A three-phase presentation is also seen, with an initial atopy/sinusitis/asthma phase, followed by an eosinophilic phase, and finally the vasculitic phase. Although DAH and GN may occur, they are much less common than in the other small-vessel vasculitides. ANCA positivity is seen less frequently than in WG, with a positive P-ANCA (or anti-MPO) seen in 35% to 75% of patients with active disease. A positive C-ANCA has been described in up to 10% of patients. Necrotizing, small-vessel vasculitis and an eosinophil-rich cellular infiltrate with necrotizing granulomas are seen histopathologically22,23.
3. Microscopic polyangiitis
MPA is considered to be a small vessel variant of polyarteritis nodosa. Distinguishing MPA from WG can be difficult because the clinical presentation, histopathologic findings, and serologic findings can be similar. Occasionally, a diagnosis of MPA is ruled out after the development of the typical clinical and serologic features of WG. The most consistent pathologic feature in MPA is a focal segmental necrotizing GN, also seen in WG, other vasculitides, Goodpasture's syndrome, and CVD1.
GN with lung involvement is seen in up to 30% of patients with MPA. In patients who develop lung disease, DAH with pathologic capillaritis is the most common manifestation. Joint, skin, peripheral nervous system, and gastrointestinal involvement are also relatively common. A positive serum P-ANCA (or MPO) strongly supports the diagnosis and can help distinguish MPA from WG. A positive P-ANCA is seen in 50% to 75% of patients and anti-MPO is seen in 35% to 65% of patients, whereas a positive C-ANCA is seen in 10% to 15% of patients. Focal, segmental necrotizing vasculitis and a mixed inflammatory infiltrate without granuloma are seen histopathologically6.
Treatment with high-dose CS and CYC or AZA is recommended. PE is of unclear benefit. As in WG, intravenous immunoglobulin may be useful for resistant cases. Factor VIIa has also been used successfully26.
4. Isolated pulmonary capillaritis
Occasional cases of DAH show no evidence of concomitant systemic involvement but show capillaritis upon histopathologic examination. Isolated pulmonary capillaritis appears to be small vessel vasculitis confined to the lungs. Some cases are associated with serum P-ANCA positivity, but most are pauci-immune. While isolated pulmonary capillaritis after treatment with all-trans retinoic acid has been observed, most cases are sporadic. Although mechanical ventilation due to respiratory failure is common, a positive response to CS and CYC has been reported. Most patients improve while on therapy, and recurrent disease is infrequent1,27.
5. Goodpasture's syndrome (anti-GBM disease)
The diagnosis of Goodpasture's syndrome, or anti-GBM disease, is reserved for cases of DAH and GN in which anti-GBM Ab (autoantibodies directed against the NC1 domain of the α3 chain of the type IV collagen in the basement membrane) appears in the serum or tissue. The typical patient is a young male smoker, although elderly persons, women, and nonsmokers are also affected. DAH may be facilitated by alveolar permeability, which is increased in most smokers28.
More than 90% of patients with Goodpasture's syndrome have serum anti-GBM Ab. In persons without circulating antibody, the diagnosis may be confirmed by lung or preferably kidney biopsy demonstrating linear deposition of antibody along the alveolar or GBM that is visible by direct IF. However, in up to 10% of patients with Goodpasture's syndrome, DAH is present without renal involvement and is identical to isolated pulmonary capillaritis, and lung biopsy with IF studies can aid in the differentiation of the two diseases. Bland hemorrhage is the most common underlying histology, but pulmonary capillaritis is sometimes seen. In such cases, kidney biopsy still shows the typical linear antibody deposition. About 30% of anti-GBM positive sera have P-ANCA (MPO) and C-ANCA (PR3) also occurs29.
Treatment of Goodpasture's syndrome requires a combination of CS, CYC or AZA, and PE. In the few cases with isolated DAH, treatment with CS alone may be effective. Cases of refractory, life-threatening Goodpasture's syndrome have been reported to respond to MMF and rituximab. The 2-year survival rate in Goodpasture's syndrome is approximately 50%1.
6. SLE and CVDs
By far the most common underlying CVD in DAH is SLE. DAH, usually caused by pulmonary capillaritis, affects 4% of patients with SLE but is life threatening when it occurs. In the majority of cases of DAH associated with SLE, GN is also present, and 80% of these cases occur in young female patients with known SLE. In immunosuppressed patients with SLE, infectious pneumonia should be excluded as the cause of DAH. DAH is distinguished from acute lupus pneumonitis by sequential BAL. Acute lupus pneumonitis presents with similar clinical and radiographic features as DAH. However, it also has the systemic symptoms typical of an SLE flare and may be the only manifestation of the initial presentation of SLE. Direct IF examination of SLE-associated DAH reveals granular deposits of immunoglobulin and complement in the alveolar interstitium and intra-alveolar blood vessels1,3.
High-dose pulse methylprednisolone (1 g given in divided doses for 3 days) can be given to those with DAH or acute lupus pneumonitis. Anecdotally, the combination of high-dose intravenous methylprednisolone and CYC is known to be the most effective combination. In refractory cases, PE has been used. The overall mortality rate for SLE-associated DAH is approximately 50% and recurrences after treatment are frequent3.
DAH has been described in rheumatoid arthritis, scleroderma, polymyositis, dermatomyositis, anti-phospholipid antibody syndrome, and mixed connective tissue disease. Treatment options are similar to those outlined for DAH in SLE1.
7. Bone marrow transplantation (BMT)
DAH occurs in approximately 5-20% of BMT recipients and the reported mortality rate ranges from 50% to 100%. Risk factors for the development of DAH in BMT recipients include older age, total body radiation, myeloablative conditioning regimens, and severe acute graft-vs-host disease. Dyspnea, fever, and cough are the most common complaints. Hemoptysis is less frequently observed in BMT recipients compared with other causes of DAH with a rate of approximately 15%. The pathogenesis has yet to be clearly established, but it has been suggested that tissue injury, inflammation, and the associated cytokine release play important roles. Lung biopsy reveals diffuse alveolar damage and DAH. The majority of patients with DAH require mechanical ventilation and CS is the mainstay of treatment, but its effectiveness is still uncertain3.
Conclusion
DAH is a clinicopathologic syndrome caused by many disorders and should be considered a life-threatening event. Common causes of DAH are systemic vasculitis such as WG, CSS, and MPA, Goodpasture's syndrome, CVD, BMT, drugs, and idiopathic conditions. DAH should be suspected in any patient with alveolar infiltrates on chest X-ray, hypoxemia, anemia, and hemoptysis. Careful attention to the medical history, physical examination, and a targeted laboratory evaluation often reveals the underlying cause. Patients suspected to have DAH should generally undergo bronchoscopy and BAL. In patients with evidence of DAH and renal involvement, kidney biopsy should be considered to identify the underlying cause and to direct therapy. A systematic approach to early recognition, establishment of diagnosis, and aggressive treatment will likely decrease the morbidity and mortality associated with DAH.

 XML Download
XML Download