

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
There are various lymphoproliferative disorders which affect the lung parenchyme. Among these, primary pulmonary lymphomas (PPLs) represent only 3~4% of extranodal non-Hodgkin's lymphoma (NHL). Pathologically, Low grade B-cell NHL accounts for 58~72% of cases of PPL1. Mucosa-associated lymphoid tissue-derived (MALT) lymphoma is a low grade B-cell extranodal lymphoma characterized by a proliferation of clonal marginal zone lymphocytes2,3 that arise from bronchial-associated lymphoid tissue4. Stimulation of bronchus-associated lymphoid tissue by antigens, smoking, inflammatory disorders, or autoimmune diseases is thought to be the initial event leading to the development of MALT lymphoma5,6. In rare cases, a nonneoplastic pulmonary lymphoproliferative disorder (p-LPD), such as lymphocytic interstitial pneumonitis (LIP) can develop into malignant, low-grade, B-cell lymphoma. A recent and realistic estimate is closer to 5% of patients with LIP acquire malignant, low-grade, B-cell lymphoma7,8. In Korea, there has been no previous report of pulmonary MALT lymphoma acquired from LIP. We describe here, a rare case of pulmonary MALT lymphoma that arose from a LIP.
Case Report
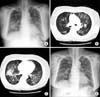
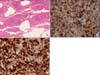
A 60-year-old woman was admitted to our hospital with cough and dyspnea. At the time of admission, her medical history included medically controlled essential hypertension, diabetes mellitus and old cerebral infarction. She was a smoker of 10 pack-years. Physical examination revealed that she had no skin lesions or neurological abnormalities. There was no lymphadenopathy in the cervical, axillary, or inguinal regions. On auscultation of the chest, a coarse breathing sound with crackles in both lower lung fields was heard. Chest radiographs and computed tomography (CT) scans showed diffuse bilateral ground-glass opacity with intralobular septal thickenings and consolidative lesions (Figure 1A~C). Laboratory investigations revealed a total leukocyte count of 7,200 cells/mm3, comprising 40.8% of neutrophils, 49.7% lymphocytes, 7.2% monocytes, 1% basophils, and 1.3% eosinophils. The level of C-reactive protein was slightly elevated to 2.39 mg/dL. The serum lactate dehydrogenase level had increased to 543 U/L (normal, 110~230 U/L). Arterial blood gas analysis in room air showed a PaO2 of 56.5 mm Hg, PaCO2 39.3 mm Hg, SaO2 90.5% and pH 7.41. Autoantibody tests were negative. Cellular analysis of the bronchoalveolar lavage fluid (BALF) revealed a composition of 25% lymphocytes, 3% eosinophils, 0% neutrophil, and 72% macrophages. Histological examination of video-associated thoracoscopic lung biopsy specimens from the left upper lobe revealed diffuse severe lymphoid cell infiltration with some plasma cells in the interstitium and alveolar walls (Figure 2A). Immunohistochemical examination revealed lymphoid cells evenly admixed with B lymphocytes and T lymphocytes, which were immunoreactive for CD79a (Figure 2B) and CD3 (Figure 2C), respectively. These findings were consistent with the diagnosis of LIP with follicular bronchiolitis components.
Our patient was treated with prednisone 1 mg/kg per day with gradual tapering for 8 months. She showed progressive clinical and radiographic improvement (Figure 1D).
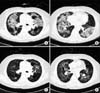
Five years later, at the age of 65 years, our patient started re-experiencing dyspnea and cough symptoms. A chest CT scan revealed diffuse bilateral ground-glass opacity with fine reticulation, traction bronchiectasis and consolidative lesions (Figure 3A, B). Pulmonary function tests revealed a reduced vital capacity (VC) of 73% predicted value, a reduced total lung capacity (TLC) of 69% predicted value and a reduced diffusion lung capacity for carbon monoxide of 49% predicted value. Cellular analysis of the BALF revealed a composition of 80% lymphocytes, 2% eosinophils, 5% neutrophil, 13% macrophages and a CD4/CD8 ratio of 0.959. Our patient was again treated with 60 mg of prednisone per day, which was gradually tapered for 6 months. This led to improvement of VC (92% of predicted value), TLC (100% of predicted) and radiographic findings (Figure 3C, D).
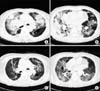
Eighteen months later, our patient suffered a relapse of dyspnea. A chest CT scan revealed diffuse peribronchial thickening, extensive consolidative lesions and nodular lesions in both lung fields (Figure 4A, B). Histological examination of percutaneous lung needle biopsy specimens revealed marked infiltration of lymphoid cells (Figure 5A). Immunohistochemical examination revealed that the lymphoid cells were diffusely positive for CD79a and B-cell markers (Figure 5B), but negative for Bcl6 (Figure 5C), CD10 and MUM-1 (Figure 5D). These findings were consistent with the diagnosis of pulmonary MALT lymphoma. Our patient was treated with 3 cycles of cyclophosphamide, hydroxydaunorubicin (doxorubicin), Oncovin (vincristine), and prednisone. Her chest radiographs (Figure 4C, D) and symptoms showed improvement. Chemotherapy was continued.
Discussion
Major nonneoplastic p-LPD, which includes lesions of the intrapulmonary lymph nodes, consists of follicular bronchitis/bronchiolitis, lymphocytic interstitial pneumonia, and nodular lymphoid hyperplasia. Such disease entities are inflammatory and reactive lesions that are often difficult to diagnose, since it is difficult to differentiate them from each other and from other neoplastic entities. Therefore, these disorders should be distinguished by clinical, radiographic, and histologic features. Radiologic findings of nodular lymphoid hyperplasia consist of well-demarcated nodules or masses. On the other hand, the CT features of LIP consist of a bilateral area of poorly defined centrilobular nodules, ground-glass attenuation, thickening of bronchovascular bundles, interlobular septal thickening, and cysts. Radiologic findings of follicular bronchiolitis are also distinct. Bilateral centrilobular and sometimes peribronchial nodules are present on a CT scan. In contrast, the nodules present in nodular lymphoid hyperplasia are generally larger, well demarcated, and often solitary with a subpleural predilection9.
Histologically, there is a confluent proliferation of lymphoid follicles in nodular lymphoid hyperplasia. LIP consists of dense polymorphous interstitial inflammatory infiltrates that diffusely expand alveolar septa. The infiltrate is composed of lymphocytes admixed with variable numbers of histiocytes and plasma cells. Reactive lymphoid follicles are more prominent and consistent in nodular lymphoid hyperplasia. In follicular bronchiolitis, numerous lymphoid follicles with associated reactive germinal centers are present around the bronchi and bronchioles10. Interstitial lymphocytes of LIP are composed predominantly of T-cells and stain positively for CD3 (pan T-cell marker). In contrast, areas of follicular bronchiolitis are composed of B-cells and stain positively for CD20 and CD79a (pan B-cell markers)10.
Diffuse bilateral ground-glass opacity with intralobular septal thickenings and consolidative lesions was observed in the radiologic studies of our patient on her first hospital admission. Histological examination of lung biopsy specimens revealed diffuse severe lymphoid cell infiltration with some plasma cells in the interstitium and alveolar walls. And there were no lymphoid follicles present in her tissue biopsy. The above mentioned polymorphous interstitial infiltration and radiologic findings were consistent with the diagnosis of nonneoplastic p-LPD such as LIP. Positive staining for CD3 (pan T-cell marker) was consistent with the diagnosis of LIP, and positive staining for CD79a (pan B-cell markers) was consistent with the diagnosis of Follicular bronchiolitis. Therefore, our patient was diagnosed with nonneoplastic p-LPD, LIP, with follicular bronchiolitis components.
Treatment of nonneoplastic p-LPD such as LIP and follicular bronchiolitis consists of steroids or other immunosuppressive agents. Therefore, we treated our patient with steroids, and in two consecutive episodes we observed an improvement of symptoms after treatment. After which, several years of remission was observed.
As seen in our patient's case, nonneoplastic p-LPD is characterized by a polymorphous infiltrate of lymphocytes. Immunohistochemical staining is positive for pan B-cell markers (CD20, CD79a) and pan T-cell makers (CD3). On the other hand, pulmonary MALT lymphoma is characterized by a fairly monomorphous infiltrate of B lymphocytes. MALT lymphoma expresses B-cell-associated antigens CD20+, CD19+, and CD79a+, and is in most cases CD5-, CD10-, CD23-, CD43+/-, cyclin D1-, and BCL6-10,11. The CT features of MALT lymphoma are, solitary nodules of poorly defined focal opacity, localized areas of consolidation with air bronchograms, ground glass opacity and small centrilobular and branching nodules12.
On her third recurrence, our patient's chest CT scan revealed diffuse peribronchial thickening, extensive consolidative lesions and newly developed nodular lesions in both lung fields. Histological findings showed a monomorphous infiltrate of lymphocytes and lymphoid cells that were diffusely positive for CD79a and B-cell markers, but negative for Bcl6, CD10 and MUM-1. So, our patient was diagnosed with pulmonary MALT lymphoma.
Stimulation of bronchus-associated lymphoid tissue by antigens, smoking, inflammatory disorders, or autoimmune diseases is thought to be the initial event leading to the development of MALT lymphoma. In rare cases, as in our patient, nonneoplastic p-LPD, such as LIP can develop into malignant, low-grade, B-cell lymphoma7,8. This is histopathologically characterized by a monomorphous infiltrate of B lymphocytes, with a predominance of smaller cell types. As stated in a previous chemokine gene and clonal analysis study, LIP may be the result of an immunologic response to a chronic antigenic stimulus. This stimulus may also provide a milieu or microenvironment for the evolution of LIP into Low-grade B-cell MALT lymphoma8. Therefore, we think that this acquirement took place in our case patient as well, as a result of chronic antigenic, inflammatory stimulation of LIP for 6 years.
Pulmonary MALT lymphoma displays an indolent but disseminated nature13. The optimal management for this disorder has not yet been clearly determined. Current treatment options are surgery and radiotherapy for localized lymphoma and chemotherapy using an alkylating agent (chlorambucil or cyclophosphamide) or purine analogue (fludarabine or cladribine) with or without the anti-CD20 monoclonal antibody, rituximab, for disseminated lymphoma14. Our patient experienced some clinical improvement after cyclophosphamide, hydroxydaunorubicin (doxorubicin), Oncovin (vincristine), and prednisone (CHOP) chemotherapy.
In conclusion, we report the first case of pulmonary MALT lymphoma that arose from LIP in Korea. The development of LIP into pulmonary MALT lymphoma is known to be caused by chronic antigenic, inflammatory stimulation. Therefore, careful follow-up is necessary for the detection of malignant transformation of nonneoplastic p-LPDs, especially LIP.

 XML Download
XML Download