

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Congenital cystic adenomatoid malformation (CCAM) of the lung is a rare developmental anomaly of the lower respiratory tract, which is characterized by multicystic lesions on terminal bronchioles. Its incidence is estimated at 1 in 25,000~30,000 pregnancies, and most cases of CCAM are found in neonates and infants with acute respiratory distress1,2. Conversely, CCAM is infrequent in adulthood. It can be accompanied by other congenital bronchopulmonary malformations, such as sequestration, bronchial atresia (BA), congenital lobar emphysema and bronchogenic cysts in children. Three types of CCAM are distinguished, and 60% of type II or type III CCAM are associated with other congenital anomalies3. Type I CCAM is the most common type, and is not generally associated with other congenital anomalies. About 52 cases of CCAM have been diagnosed in adults, but it is thought that only one case has been reported of CCAM associated with BA4.
We present this case because of its infrequent occurrence in middle-aged patients and its association with another congenital pulmonary anomaly, BA.
Case Report
A 54-year-old female was referred to our respirology specialty clinic for evaluation of a small amount of blood-tinged sputum. She had had a chronic cough accompanied by small amounts of sputum for 10 months, and had noticed intermittent small amounts of blood-tinged sputum in the last few days. One week prior to her hospital visit she had visited a local clinic and had been diagnosed as bronchiectasis on the basis of simple radiographs. She had been treated with antibiotics for 7 days, but her symptoms had not been relieved. About 20 years previously she had been diagnosed with pulmonary tuberculosis and had been treated successfully, and had had no further respiratory symptoms until 10 months before. She had had a history of spine operation for her back pain 14 years prior to her admission but no other underlying disease had been diagnosed. She had never had upper respiratory infections or pneumonias requiring antibiotic treatment, nor had she been hospitalized. To her knowledge she had never undergone chest image before visiting the local clinic. The patient was a lifetime non-smoker, and at baseline was active and healthy. She did not complain of chest pain or dyspnea, or any limitation of her activity level. Vital signs on admission were stable, and her chest was completely clear to auscultation, without wheezes, rhonchi, or rales. The rest of her examination was unremarkable. The spirometry test was notable for a forced expiratory volume in 1 second (FEV1) of 1.40 L (71% predicted), forced vital capacity (FVC) of 3.53 L (88% predicted), and FEV1/FVC ratio of 69%. The flow-volume loop suggested mild obstruction, and lung volumes were not measured at the time.
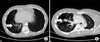
Simple chest radiographs revealed a patchy opacity with multiple thin-walled cystic structures in the right lower lobe of the lung field (Figure 1). A computed tomography (CT) scan of the chest showed multiple large air-filled cystic lesions in the right lower lobe, which measured 12.0×11.0×5.5 cm3 in aggregate and were primarily located in the right lower lobe. At its perimeter were many small, fluid-filled spaces, the largest of which measured 1.5×1.0×1.0 cm3, and there was some calcification. These radiologic findings were consistent with type I CCAM (Figure 2A). Moreover, an oval-shaped opacity in the distal right middle lobe bronchus was surrounded by a low attenuation in the right middle lobe and there was no clear connection between the lesion and the tracheobronchial tree (Figure 2B); this was suggestive of BA. The other lung parenchyma was normal.
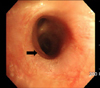
On a flexible bronchoscopic examination, the opening of the right middle lobe bronchus was not seen and there was no endobronchial lesion (Figure 3). Based on radiologic and bronchoscopic findings, the patient seemed to have CCAM in the right lower lobe and BA in the right middle lobe.
To diagnose and treat this lesion, the patient underwent a right middle lobectomy and a medial basal segmentectomy of the right lower lobe via a right posterolateral thoracotomy. The cystic malformation was excised en bloc along with the right lower lobe. The operation was completed without complications. On gross inspection, the specimen showed multilobular cystic lesions ranging in size from 0.3 to 1.8 cm in diameter, separated by grossly unremarkable pulmonary parenchyma. The diameter of the whole cystic lesion measured 12.5×11.0×5.2 cm3, and there was yellowish white mucous and necrotic material in the medial basal segment of the right lower lobe. Histologically, the right lower lobe was seen to contain a multilocular cystic lesion lined by stratified ciliated columnar epithelium, with underlying cartilage and mucus-secreting glands. There was marked infiltration of neutrophils, lymphocytes, and plasma cells in the cyst wall, with focal calcification and a proliferation of small blood vessels. These findings were consistent with type I CCAM (Figure 4A). The bronchus of the right middle lobe of the lung was cystically dilated and lined by pseudostratified ciliated columnar epithelium. The ciliated bronchus contained mucus material, and the bronchial wall contained underlying cartilage and mucous glands with few calcifications. The alveolar spaces distal to the ectatic bronchus were dilated and there was septal smooth muscle proliferation (Figure 4B). These findings favor a bronchocele. All 24 lymph nodes were unremarkable. Since removal of the anomalies the patient has been followed up in the outpatient-clinic for 2 years without any complications.
Discussion
Most cases of CCAM are diagnosed by routine prenatal ultrasound or in the immediate neonatal period, and CCAM that develops in adulthood is perhaps the rarest presentation. Only about 50 cases of CCAM in adulthood have been reported in the English language literature. There have been fewer than 5 cases of CCAM in persons over 50 and we believe that the present patient is the oldest known female with CCAM. The association of type I CCAM and BA is extremely rare and just one case has been reported: in an adult patient in Italy4. The present case is therefore the second case of CCAM with BA in adults, and the first presentation in a non-Caucasian.
About 25% of congenital cystic lung lesions are diagnosed as CCAM by ultrasonography in the prenatal period. Up to 71% of such cases are asymptomatic at birth, and spontaneous regression has been reported in as many as 76% of cases without prenatal intervention5; approximately 85% of cases are diagnosed in the first 2 years of life and presentation is only rarely delayed until adulthood, as in our patient6. Although almost cases of CCAM are without symptoms and found incidentally, a few cases have dyspnea, hemoptysis, spontaneous pneumothorax, recurrent respiratory tract infections, pneumonia or lung abscesses7. The present patient had had a chronic cough for 10 months, and blood-tinged sputum.
Since Stocker in 1975 classified CCAM into three subtypes based on clinical, gross and histological features, his classification has been widely used2. Type I is the most frequently encountered (50~70% of cases), and presents with single or multiple large cysts (>2 cm in diameter). The largest cysts are lined by ciliated pseudostratified columnar epithelium and often have a papillary or polypoid appearance. Mucus-producing cells are present in approximately one-third of cases, whereas cartilage in the wall is rare. Relatively normal alveolar ducts, alveolar saccules and alveoli can be seen between the cysts. Type II lesions (approximately 40% of cases) are characterized by multiple small cysts. Mucous cells and cartilage are absent, although striated muscle fibers may occasionally be seen. Type III lesions are rare (3% of cases). They have a solid appearance made up of bronchiole-like structures lined by ciliated cuboidal epithelium and separated by masses of alveolar-sized structures lined by non-ciliated cuboidal epithelium. About 4~26% of cases are associated with other congenital abnormalities and this may be up to the time of developing anomaly at the stage of embryonic development and about half the patients with type II and III CCAM have associated neonatal anomalies8. Unlike the two other types of CCAM, type I CCAM is rarely associated with other anomalies and has an excellent prognosis. Cases arising in adulthood are rarely associated with other congenital anomalies.
Radiographic finding can help to diagnose CCAM, but simple chest radiography is nonspecific, and consolidations have been described that were indistinguishable from lobar pneumonia, lung abscesses or pneumatoceles. Sometimes they may not be visible on chest X-rays9. CT is the best method for identifying CCAM lesions. In the present patient, the CT findings included a large lesion with multiple cystic areas containing secretions. Type I CCAM is characterized by large cysts (>1 cm in diameter) on imaging. The lesion in CCAM is usually unilateral and sublobar or lobar inside the right lower lobe or left lower lobe3 but occasionally it can be multilobar or even bilateral with no special predilection for any lobe. However histological confirmation is needed because of the wide range of radiologic expression of CCAM and the difficulty of making a correct preoperative diagnosis. Radiologic differential diagnosis must be made with pulmonary sequestration, bronchiologenic cyst, cystic bronchiectasis, diaphragmatic hernia, and infected tumor8,9. In our patient, histological examination revealed multiple cysts lined by pseudostratified ciliated columnar epithelium, and the scattered mucous cells and pulmonary parenchyma around the cysts and surrounding the lesion were normal. These findings are consisted with type I CCAM.
It is believed that CCAM is generally associated with the development of neoplasms. Even in asymptomatic young patients, transitions into bronchioloalveolar carcinoma10, pleuropulmonary blastoma and rhabdomyosarcoma11 have been seen by chance. It is thought that these changes are caused by the presence of metaplastic mucous cells, primitive mesenchymal cells and differentiated but poorly organized striated muscle fibers. The treatment of CCAM should be the same as that for bronchial non-small-cell cancer. Anatomic resection with extended lymphadenectomy is the treatment of choice, and the prognosis after radical excision seems to be excellent. As we were concerned about the possibility of malignant transformation we removed the anomaly surgically.
BA typically results from congenital focal obliteration of a proximal segmental bronchus with distal structures remaining intact. It is a more common congenital anomaly than CCAM, and more than 100 cases have been reported in adults. BA is usually diagnosed in the second or third decade of life. It seems that the disorder occurs mainly in males, with a prevalence of 1.2 cases per 100,000 males12. About half to two thirds of the reported cases are asymptomatic before diagnosis, and this explains their late detection. Half of BA cases in children have associated anomalies: distal bronchiectasis, bronchogenic cyst, anomalous branching of bronchial trees and vascular structures13, but cases of BA accompanied by type I CCAM are rare in adults4. Almost all cases of BA are diagnosed by radiological methods such as chest radiography or chest CT rather than bronchoscopy or surgical procedures. Bronchoscopy can identify a blind-ended bronchus, but it can also reveal no abnormality. Also, the absence of a segmental or sub-segmental bronchus may be considered normal anatomical variability of the bronchial tree rather than BA, in the absence of the characteristic radiographic features. Hence, in the majority of cases, congenital BA remains a radiological diagnosis. However, some workers have suggested that similar radiographic findings could be obtained in serious disorders such as lung cancer or bronchial adenoma. The role of bronchoscopy is to exclude these disorders and demonstrate the patency of the central bronchi, especially in doubtful cases. But since BA has low malignant potential, surgical removal is not generally recommended.
There has been only one previous case of type I CCAM with BA4. In that case the patient was younger (34-year-old) and had had recurrent pleuritic chest pain and many episodes of lower respiratory infection before visiting the clinic. She underwent bronchoscopic biopsy for diagnosis, and surgery was not performed. Her lesions were located in various lobes of the right lung, as in our patient, but the CCAM was in the apical segment of the right lower lobe and the BA in the posterior segment of the right upper lobe. The patient was examined regularly because of the malignant potential of CCAM. In our case the patient was older and had mild symptoms such as cough and sputum but without signs of infection. To obtain a differential diagnosis and because of the potential malignancy, the lesions in the right middle lobe and medial basal segment of the lower lobe were removed, and the diagnosis was established as CCAM with BA. There was no evidence of malignancy and the patient recovered without complications.
CCAM of the lung is a rare congenital anomaly that typically affects neonates. There have been few cases in adulthood and type 1 CCAM is rarely associated with other respiratory anomalies. However further evaluation is needed in patients with insidious symptoms like chronic cough or cystic lesions seen by radiography, since the lesion may be an undetected congenital anomaly with high malignant potential. Chest CT scan and surgical biopsy must be considered for diagnosis and treatment of these lesions. This is the first report of CCAM and BA in a non-Caucasian adult; the lesion was removed for differential diagnosis and treatment.


 XML Download
XML Download