

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Acute lung injury (ALI) and its severe form of acute respiratory syndrome (ARDS) are characterized by acute hypoxemic respiratory failure, low pressure pulmonary edema, and massive neutrophilic infiltration in the lung1. The fundamental pathogenesis that initiates and intensifies this intractable inflammatory process has not been elucidated. Phospholipase A2 (PLA2) has long been known to participate in ALI, and various studies have been carried out to develop new therapeutic modalities for acute lung injury2,3. Until now, however, the results are discouraging and the heterogeneity of etiologies increases the difficulties to explain the exact pathogenesis of ALI4.
Fat embolism syndrome (FES) is one of the etiologies causing ALI and oleic acid in the bone marrow or other part of the body is the main source of acute inflammatory lung injury in the FES5. OA not only directly injures endothelial cells of the lung6, but also initiates inflammatory reaction through the activation of PLA27.
In spite of the unsuccessful outcome to develop new therapeutic drugs against ALI, the recent discovery of new types of PLA2s and the novel knowledge on these new PLA2s have prompted studies on PLA2 expecting to discover new therapeutic modalities of ALI.
Though PLA2s have been studied widely in conjunction with ALI8 and its new therapy, the efforts have been in vain still. However, in FES-induced ALI, cytosolic PLA2 (cPLA2) might play a crucial role for the initiation of acute inflammation in the lung, as cPLA2s are abundant in the lung and mononuclear phagocytes are enriched with cPLA29. Besides, cPLA2 mediates arachidonic acid-induced tissue injury which comprises the activation of NADPH oxidase and inflammatory reaction irrespective of free radicals10. In OA-induced ALI, neutrophils play a crucial role along with its accumulation and migration to the lung provoking epithelial and endothelial injury by oxidative stress and release of proteases11.
Recently, fluoroquinolones were discovered to have immunomodulatory functions, especially anti-inflammatory effects12,13 by suppressing the migration of phagocytes in some clinical conditions. Considering the possibility that moxofloxacin's immunomodulating effects are linked to cPLA2 activity, effects of moxifloxacin on OA-induced ALI were tested in conjunction with the cPLA2-activated neutrophilic oxidative stress and the activation of TNFα and COX-II in the lung.
Materials and Methods
1. Experimental animals and reagents
Male Sprague-Dawley rats were purchased from Sasco Company (Sasco Korea, pathogen free, 250~300 g). Goat anti-human cPLA2 polyclonal antibody and anti- COX-II antibody were purchased from Santa Cruz Biochem (Santa Cruz, CA, USA). Anti-TNF-α-polyclonal antibody was obtained from Cell Signaling (Danvers, MA, USA). L-α-dipalmitoyl-[2-palmitoyl-9,10-3H(N)-phosphatidylcholine was obtained from Dupont NEN Research Products (Boston, MA, USA). Moxifloxacin hydrochloride was graciously provided by Bayer (Bayer Leverkusen, Germany). Not otherwise mentioned, all other reagents were purchased from Sigma Aldrich Company (St. Louise, MO, USA).
3. Treatments
Rats were divided into three treatment groups
1) Sham group: rats were given 300µL of 10% bovine serum albumin intravenously via femoral vein; 2) Oleic acid control group (OA group): rats were given 30µL of oleic acid with 270µL of 10% bovine serum albumin intravenously; 3) Moxifloxacin group (MO group): rats were given moxifloxacin hydrochloride (100 mg/kg) with 30µL of oleic acid in 270µL of 10% bovine serum albumin. Moxifloxacin hydrochloride was given intraperitoneally just after the injection of oleic acid.
4. Bronchoalveolr lavage (BAL)
BAL was performed 5 hours after oleic acid injection. The rats were anesthetized with enflurane and tracheotomized and cannulated. BAL was performed with 8.0 mL of PBS for each rat and approximately 6.0 mL of BAL was recovered. BAL fluid was collected into 10 mL sterile tubes, centrifuged to collect cell free supernatant and stored at -20℃ for protein assay. The sediments were resuspended with PBS for counting of leucocytes.
5. BAL protein
Determination of BAL protein was performed as previously described by Brown et al.15 with modification. Stored BAL supernatant was used for protein assay.
6. BAL neutrophil count
TenµL of resuspended sediment solution was applied on hemocytometer and leukocytes were counted. Around 100µL of BAL suspension was cytospinned and slides were prepared before Wright staining. Percentage of neutrophils was calculated by differential count and total numbers of neutrophils were calculated.
7. Lung myeloperoxidase (MPO) assay
The MPO activity (U/g of wet lung) was determined to estimate the accumulation of neutrophils in the lung. The assay was performed as described by Goldblum et al.16 with modification. Lungs were weighed and homogenized with Polytron homogenizer (Polytron, Switzerland) in 4.0 mL of 20 mM PBS at pH 7.4. An aliquot was mixed with an equal volume of 1% hexadecytrimethyl ammonium bromide in PBS and rehomogenized for 90 s in ice cold water. Then it was centrifuged 15,000 g for 5 minutes at 4℃. The supernatant (0.1 mL) was added to 2.9 mL of PBS (50 mM potassium phosphate, pH 6.0) containing 0.0005% H2O2 and 0.168 g/L of o-dianisidine. The samples were analyzed for change in absorbance on spectrophtometer at 460 nm over 3 minutes. MPO activity was expressed as U/g of wet lung by multiplying 13.5 to the changes of absorbance/min/g of wet weight tissue.
8. Determination of lung malondialdehyde (MDA); thiobarbituric acid reactive substance (TBARS) assay
TBARS assay was carried out as described previously17 to measure lipid peroxidation as a marker of ALI. The same supernatant used for MPO assay was used. Aliquots of supernatant were mixed with equal volume of 3.5% sodium dodecyl sulfate (SDS) and 0.13 mL of 100% acetic acid in 15 mL-glass centrifuge tubes. The pH was adjusted to 3.5 with 1 N NaOH. Five mL of 0.6% thiobarbituric acid was added to each tube and they were heated in boiling water bath at 95℃ for 30 minutes. After cooling, 1.5 mL of n-butanol/pyrimidine (15:1) mixture was added to each tube and vortexed vigrously for 30 s followed by centrifugation at 4,000 rpm for 10 minutes. The absorbance of top layer of each sample was measured by spectrophotometer at 525 nm.
9. Determination of cPLA2 activity
cPLA2 activity was determined by the hydrolysis of radioactively labelled phospholipid (3H-dipalmitoylphosphatidylcholine, 3H-DPPC) as previously described by Katsumata et al.18 with modification. Lung tissues were homogenized in a polytron in ice-cold buffer (1:5, wt/vol) containing Tris-HCl (pH 7.6), EDTA (1 mM), EGTA (1 mM) and DTT (5 mM) to abolish sPLA2 activity. The phospholipid substrate solution (pH 9.0) was composed of 3H-DPPC, lecithin (0.1 mM), glycine (100 mM), deoxycholate (2.5 mM), BSA (10 g/L) and 1.75 mM ethanol. The reaction was initiated by the addition of 100µL of lung homogenate to the phospholipid substrate solution and incubated at 37℃ in a shaking water bath for 60 minutes. Crotalus adamanteus PLA2 was used as a positive control. The reaction was stopped by adding 200µL of 5% Triton X-100 containing 200 mM EDTA and 5 g of anhydrous Na2SO4. For the extraction of hydrolysis product, 5.0 mL of 0.1% glacial acetic acid-hexane was added to each sample. The extract was scanned with β-scintillation counter. 1 unit of PLA2 activity was defined as 1µM production of fatty acid per minute.
10. Lung histological preparations
Lungs were fixed by inflation with paraformaldehyde (pH 7.4, 4%) at pressure of 20 cm H2O and sliced before putting into paraformaldehyde, then degassing and fixed. Tissue slices were stained with hematoxylin-eosin for examination of lung histology.
11. Immunohistochemistry of cPLA2 in rat lung tissue
Microtomer sections were subjected to staining for fluorescence and diaminobenzidine (DAB) for cPLA2 in rat lung tissues. Goat anti-human-cPLA2 polyclonal was used as first antibody. The antibody complex was visualized by propidium iodide (fluorescense) and DAB staining.
12. Protein expression of TNFα and COX-2
Proteins of TNFα and COX-2 were identified in homogenized lung tissue (100µg of protein) using standard western blotting. Specific polyclonal antibodies, anti-TNF and anti-COX-II were diluted 1:1,000 (Tris buffer, Saline Tween 20 [TBST] buffer with 0.1% BSA).
Results
1. Effects of moxifloxacin on lung leak
The assay of BAL protein revealed the protective effect of moxifloxacin on lung leak in rats. As shown in Table 1, OA had increased the protein content (mg/two lungs) in BAL of rats given OA, as compared with that of sham-treated rats (p<0.001). Moxifloxacin had decreased the protein content in BAL of rats given OA (p<0.01).
2. Effects of moxifloxacin on the accumulation and migration of neutrophils in the lungs of rats given the OA
As noted in Table 2, moxifloxacin reduced the accumulation of neutrophils in the interstitium of the lung and suppressed the migration of neutrophils into the alveoli of the OA group (p<0.001). As expected, the OA caused dramatic mobilization of neutrophils manifested by the drastic increase of lung MPO (p<0.001). The accumulation of neutrophils in the lung interstitium caused the migration of neutrophils into the alveoli as a process of alveolar flooding. Moxifloxacin was effective to decrease the effects of OA on the mobilization of neutrophils (p<0.001).
3. Estimation of protective effects of moxifloxacin by TBARS assay
As an index of lung injury, the TBARS in the lung was measured. The MDA content (nmol/g of lung) was higher in the lungs of OA given rats compared with that of sham treated rats (p<0.001). The MDA content in the lungs of moxifloxacin given to rats was lower than that of the rats given OA reflecting the protective effect of moxifloxacin (p<0.001) (Table 3).
4. cPLA2 activity in the lung tissue
As shown in Table 4, the cPLA2 activity was higher in the lungs of OA-treated rats compared with that of sham-treated rats (p<0.001). The cPLA2 activity was lowered in the lung tissue by moxifloxacin in OA-treated rats (p<0.05).
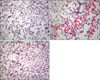
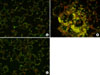
5. Lung histology and cPLA2 immunohistochemistry of lung tissue
While examining the histopathology of the lung of rats given OA, many phagocytes, mainly neutrophils, were observed. Perivascular cuffing and alveolar flooding were also observed, as these were the results of the OA injection (Figure 1A and B). Moxifloxacin ameliorated these inflammatory reactions dramatically, even if the remnants of migrated phagocytes, mainly alveolar monocyte/macrophages remained (Figure 1C). Along with these histopathological changes, the immunohistochemistry revealed the inhibitory effects of moxifloxacin on cPLA2 in the lung. The DAB and fluorescent staining of cPLA2 in the lung demonstrated the diminished expression of cPLA2 by moxifloxacin (Figures 2, 3). Note the deep staining of cPLA2 in the intra-alveolar neutrophils with DAB in the OA-treated rats. Interestingly, this phenomenon disappeared by moxifloxacin almost completely. In the lung tissue, the fluorescent staining of the alveolar septa was shown in OA-treated rats, and again, moxifloxacin suppressed this phenomenon (Figure 3).
6. Western blotting of TNFα and COX-2 in the lung tissue
The immunoprecipitation of TNFα and COX-2 was carried out to estimate the anti-inflammatory effects of moxifloxacin associated with the pro-inflammatory cytokine and rate-limiting enzyme of inflammation. The OA increased the expression of TNFa and COX-2, which was normalized by moxifloxacin (Figure 4).
Discussion
Moxifloxacin is a fluoroquinolone that acts against gram positive and gram negative bacteria19. It has a dual role of antimicrobial and protective anti-inflammatory effects in clinical conditions20.
In the present study, the property of immunomodulating effect of moxifloxacin was evaluated on OA-induced ALI in terms of protection from neutrophilic inflammatory reaction. As expected, moxifloxacin was shown to exert a protective effect on OA-induced lung leak and to suppress the accumulation and migration of neutrophils in the lung.
The OA is one of the derivatives of fatty acids and is responsible for acute inflammatory injury of the lung in FES, that is, OA induces lung leak, accumulation of neutrophils, and oxidative stress leading to ALI in experimental animals and human beings21. Moxifloxacin was effective to suppress lung leak by the decrease of neutrophilic oxidative stress or neutrophil-induced tissue injury evidenced by reducing lung MPO, number of BAL neutrophils, MDA content in the lung of rats given OA. Specifically, the lower content of MDA in the lung by moxifloxacin signifies the anti-oxidative stress effect of moxifloxacin.
According to Shalit and associates13, moxifloxacin was effective to inhibit the migration of neutrophils on endothelial cells resulted in the decrease of cellular injury. The results in this experiment are consistent with their report.
In addition, moxifloxacin's anti-inflammatory effects were demonstrated prominently in lung histology. Although the mechanism is obscure, the decreased activity of cPLA2 and the expression in the lung might be one of the clues to understand the anti-inflammatory property of moxifloxacin.
Until recently, the relationship between the role of PLA2 and the immunoregulatory effect of moxifloxacin has not been elucidated. Since the role of cPLA2 on ALI is predominant in provoking acute pulmonary edema, the decrease of the pulmonary edema by moxifloxacin might be originated from the down regulation of cPLA2. Because the activation of cPLA2 by OA was evident and the activation of cPLA2 would be followed by the synthesis of pro-inflammatory lipid molecules22, it is natural to envisage that the moxifloxacin's anti-inflammatory effect was derived from the inhibition of cPLA2.
In western blot analysis, the expression of TNFα was stimulated by OA, and moxifloxacin depressed the increased expression of TNFα by OA. According to the classical process of the inflammation, the activation of cPLA2 could be initiated by the increased expression of TNFα, one of the pro-inflammatory cytokines which induced inflammatory reaction in ALI23. The decreased expression of TNFα by moxifloxacin might subsequently decreased the cPLA2 activity in the lung and suppressed the expression of cPLA2 in the lung and neutrophils.
One more interesting result was that the OA increased expression of COX-2, one of the rate-limiting enzymes of inflammation. The increased expression of COX-2 by OA was lowered by moxifloxacin, which implied the anti-inflammatory property of moxifloxacin in ALI. Moxifloxacin seemed to block the downstream of the inflammatory process by disintegration of positive feedback loop of neutrophilic oxidative stress24.
In other words, moxifloxacin suppressed secretion of TNFα from phagocytes which down regulated cPLA2 and was followed by the decreased expression of COX-2. From the inflammatory reaction's point of view, the block or suppression of pro-inflammatory cytokine presumed to lower the cPLA2 activity in the lung by moxifloxacin.
Conclusively, though further studies are to be desired to understand the precise mechanism of the effects of moxifloxacin on FES-induced ALI, based on the present study, I would like to suggest that the suppression of cPLA2 by moxifloxacin might be protective against injurious neutrophilic oxidative stress in rats.






 XML Download
XML Download