

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Common variable immunodeficiency (CVID) is a primary immunodeficiency characterized by B cell dysfunction. CVID generally comprises antibody deficiencies that present in either late childhood or, more typically, early to mid adulthood1. The diagnosis is based on decreased serum immunoglobulins and a failure to produce antigen-specific antibodies in response to vaccinations or infections2. We report a CVID case of a mid-aged woman who had presented with recurrent severe pneumococcal pneumonia.
Case Report
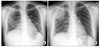
A thirty three-year-old woman admitted our hospital with chilling sense, myalgia and dyspnea. Before admission, she felt chilling and myalgia for 5 days and shortness of breath for 3 days. She suffered recurrent rhinorrhea, cough, and phlegm during last ten years. She was diagnosed chronic rhinosinutis 5 years ago, and iron deficiency anemia three months ago. Five months before admission, she got myalgia and chilling sense for several days and had taken antibiotics at private clinic. Three months ago, she admitted this hospital because of bilateral pneumococcal pneumonia with bacteremia (Figure 1A). At that time, she received cefotetan (2 g, bid, iv) with klarithromycin (500 mg, bid, oral) for 14 days. With full recovery, she discharged. She had received vaccinations as scheduled except pneumococcal vaccine. She had no regular medication except iron pill. Her family history was not remarkable. She has been married and housewife.
She appeared chronically ill and showed frequently coughing mixed with purulent sputum. There were no gastrointestinal symptoms. Her height was 160 centimeters and weight was 50 kilograms. Her blood pressure was 120/80 mm Hg, pulse rate 88 beats per minute, respiratory rate 18 per minute, and body temperature 37.6℃. There was no facial anomaly. Cervical and axillary lymph nodes were not palpable. On lung sound, inspiratory coarse crackle was heard on her right upper thorax. There were no clubbing fingers.
Initial white cell count was 13,400/mm3 with 77% segmented neutrophils and 18% lymphocytes. No abnormal blood cells were on peripheral blood smear. Hemoglobin was 11.7 g/dL, Hct 36.6% , MCV 82 fL (normal range, 81~101), MCH 26.2 pg (normal ragne, 26.2~34.2) and platelets were 213,000/mm3. In urinalysis, no protein and red blood cells were detected. In blood chemistry, BUN was 11.5 mg/dL, creatinine 0.57 mg/dL, uric acid 3.0 mg/dL, total bilirubin 0.6 mg/dL, AST 14 IU/L, ALT 16 IU/L, alkaline phosphatase 297 IU/L, total protein 6.5 g/dL, albumin 4.3 g/dL and globulin 2.2 g/dL. Serum iron was 6µg/dL (normal range, 50~170), total iron binding 305µg/dL (normal range, 262~474) and ferritin 65 ng/mL (normal range, 5~148). Serum protein electrophoresis showed a markedly decreased gamma globulin 0.08 g/dL (normal range, 0.7~1.7) (Figure 2). Precipitated lines about Ig A, G, and M in serum immunoglobulin electrophoresis were vague. Each serum immunoglobulins were decreased as a whole: serum Ig A 43 mg/dL (normal range, 800~1,800), Ig G 1 mg/dL (normal range, 90~450), and Ig M 19 mg/dL (normal range, 65~333). Her blood type was A and Rh (+). Serum iso-hemagglutinin tests anti-A and anti-B on her red blood cells were negative. Anti-tetanus antibody was not detected on second admission. Anti HIV antibody was negative. In lymphocyte subset flow cytometry, CD4+ T cells were 896/mm3 (normal range, 700~1,100), CD8+ T cells 979/mm3 (normal range, 500~900) and CD19+ B cells 351/mm3 (normal range, 200~400).
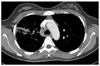
Her chest x-ray showed wedge shaped consolidation in right upper lung field (Figure 1B). Chest computed tomography revealed pneumonic consolidation and ground glass opacities of right upper and lower lung fields with reactive mediastinal and right supraclavicular lymphadenopathies (Figure 3). Transthoracic echocardiogram showed normal cardiac function and no evidence of endocarditis. Esophagogastroduodenoscopy and colonoscopy were normal. Abdominal and pelvic computed tomography showed normal findings.
Her urinary pneumococcal antigen was positive and Streptococcus pneumoniae grew in the sputum and blood cultures. After intravenous or oral moxifloxacin (400 mg, qd) for two weeks, she was able to discharge because of clinical and radiological improvement. Her illness was common variable immune deficiency. She has been treated by intravenous immunoglobulin 400 mg/kg monthly. Up to 1 year follow up, there was no serious infection event.
Discussion
CVID has variable clinical manifestations, the most common being recurrent bacterial infections caused by encapsulated bacteria such as Streptococcus pneumoniae, Haemophilus influenzae. Bacterial infections commonly involve the paranasal sinuses and respiratory tract2. As CVID is considered as genetic disease, it is the sporadic pattern and relatively late onset. The mean age at diagnosis is between 25 years and 45 years of age. This disorder results from failed B-cell differentiation, with impaired secretion of immunoglobulins3. A lot of defects of T-cell function and deficits in the memory B-cell pool have been identified, but the underlying cause of this defect remains unknown. Therefore, the hallmark of CVID is hypogammaglobulinemia, and the standard treatment is IV immunoglobulin replacement. On the other hand Ig A, Ig G and/or Ig M levels should be less than two standard deviations below the mean for age-adjusted standardized reference, and reduced levels of at least two Ig isotypes2. To confirm the diagnosis of CVID, there were inadequate antibody responses to pneumococcal vaccine and tetanus toxoid or absent isohemagglutinins4. She did not receive tetanus vaccine on admission. Unfortunately we did not evaluate the anti-Tetanus titer for response of vaccination. Our patients also showed markedly decreased gamma globulin fraction in serum protein electrophoresis and immunoglobulins despite of negative Anti Tetanus Ab. We observed slightly enlarged mediastinal and right supraclavicular lymph nodes. There were normal sized spleen and abdominal lymph nodes. We considered as reactive lymph node enlargements, so did not perform mediastinoscopic lymph node biopsy. Throughout clinical and radiological examinations, we should evaluate malignancy such as lymphoma. Also there is an increased risk of malignancy in CVID. Chest commuted tomography should be followed as recommended. We excluded other causes of hypogammaglubulinemia as guided5. There was no drug exposure and evidence of chromosomal abnormalities. She had no conditions of excessive loss of immunoglobulin such as nephrosis, severe diarrhea. Because of female patient, normal CD19+ B cells and T cells counts, we could exclude X linked aggammaglobulinemia and severe combined immnunodeficiency.
There were three CVID case reports in Korea. One was a seventeen years old girl who had presented continuous infectious events from early childhood6. The second case was a 10-year old boy who diagnosed with CVID and a fatal measles7. The other was a twenty years old woman who was diagnosed by granulomatous liver disesase but her recurrent infectious episodes had started 14 years ago8. But our case was somewhat typically presented in a mid-thirty aged woman whose infections had initiated 5 years ago.
Intravenous immunoglobulin (IVIG) treatment in addition to early diagnosis was thought as important prognostic factor in CVID. IVIG is effective and is currently the mainstay of therapy for CVID9. Intravenous immunoglobulin also reduces the incidences of pneumonia and serious recurrent bacterial infections and prevents chronic lung disease and enteroviral meningoencephalitis10.
Besides recurrent infections, CVID patients have an increased tendency to develop autoimmunity, lymphoproliferative disease and malignancies. Although these disease complications cause severe morbidity, the enormous heterogeneity in the clinical presentation of CVID induced delays in recognizing its illness and complications. As this case presented, CVID should be suspected, if any patient showed recurrent infections, especially of the upper or lower respiratory tract. After diagnosis of CVID, the complications and associated conditions should be followed up.

 XML Download
XML Download