

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Congenital unilateral pulmonary artery hypoplasia or agenesis without congenital intracardiac anomalies (isolated unilateral pulmonary artery hypoplasia [UPAH] or unilateral pulmonary artery agenesis [UPAA]) is a rare disease in elderly adults1. Diagnosis is often delayed in patients with isolated UPAH or UPAA because symptoms are absent due to abundant systemic collaterals to the affected lung or because UPAH or UPAA mimics other pulmonary diseases such as infectious sequelae2.
Computed tomography (CT) and angiography are important in the diagnosis of UPAH and UPAA. More recently, however, magnetic resonance imaging (MRI) has been used to provide more accurate visualization and identification of accompanying abnormal anatomy of the heart and great vessels3-5.
We encountered a patient with isolated UPAH and retrograde blood flow from the systemic collaterals to the left pulmonary artery via right pulmonary artery, as determined by MRI. To our knowledge, there have been no reports on the use of this non-invasive method to detect retrograde blood flow and our patient is the oldest to date with isolated congenital UPAH.
Case Report
A 68-year-old man was admitted for dyspnea on exertion (DOE) of 23 years' duration. Twenty-one years earlier, he had been diagnosed with right tuberculous pleurisy and treated with anti-tuberculous medications for 10 months. His DOE did not improve. Eight years previously, on first visiting our hospital, he was diagnosed with obstructive lung diseases, consisting of chronic obstructive pulmonary disease (COPD) and combined restrictive lung disorder due to pleural adhesion caused by the previous tuberculous pleurisy. He was treated with medications such as bronchodilators and showed some improvements in symptoms for a few years. Despite these medications, however, his DOE worsened, beginning six months before his first admission.
His previous medical history included hypertension, but no other diseases such as diabetes or hepatitis. He denied having a childhood pulmonary infection. He had no specific family history, but was an ex-smoker with a smoking history of 38 pack-years. Physical examination showed that his blood pressure was 120/69 mm Hg, his pulse rate was 75 per minute, his respiratory rate was 20 per minute and his body temperature was 36.5℃. He was alert, his general condition was good, and he did not have an acute illness. Auscultation showed inspiratory crackles in the right posterior lower lung field. His heart sound was regular without murmurs, however, he had severe finger clubbing.
Laboratory findings showed that his white blood cell count was 9,600/mm3 (neutrophil 59.1%), hemoglobin level was 17.2 g/dL and platelet count was 298,000/mm3. Blood chemistry analysis revealed a glucose concentration of 94 mg/dL, a creatinine concentration of 1.1 mg/dL, a blood urea nitrogen concentration of 17 mg/dL, a total protein concentration of 7.3 mg/dL, and an albumin concentration of 3.9 mg/dL. His aspartate aminotransferase was 21 IU/L, alanine aminotransferase was 23 IU/L, alkaline phosphatase was 54 IU/L, total bilirubin was 0.9 IU/L. His arterial blood gas analysis showed that pH was 7.475, pCO2 was 26.9 mm Hg, pO2 was 63.8 mm Hg and bicarbonate was 19.9 mm Eq/L. His alveolar-arterial oxygen tension gradient was 52 mm Hg.
Pulmonary function tests (PFT) at his first visit showed that his forced vital capacity (FVC) was 2.99 L (70% of predicted), forced expiratory volume in one second (FEV1) was 1.90 L (63% of predicted), FEV1/FVC was 64%, total lung capacity 4.76 L (77% of predicted), and diffusing capacity for carbon monoxide (DLCO) was 13.9 mL/mm Hg/min (63% of predicted), indicating an obstructive and restrictive pulmonary disorder with decreased diffusion capacity. After 8 years later, at his first admission, his FVC was 2.97 L (72% of predicted), FEV1 was 1.93 L (68% of predicted), FEV1/FVC was 65% and DLCO was 11.2 mL/mm Hg/min (55% of predicted).
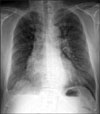
Chest radiography showed a slightly smaller right lung with decreased pulmonary vasculature and mild emphysematous changes in both lung fields. We observed fine linear opacities in the right lung and blunting of the right costophrenic angle, indicative of previous pulmonary tuberculosis and tuberculous pleurisy (Figure 1).
Transthoracic echocardiography (TTE) showed normal LV contractility, chronic pressure overload of the right ventricle and severe resting pulmonary hypertension. The TR Vmax was 4.2~4.7 m/sec. Transesophageal echocardiography (TEE) showed no abnormalities in valvular morphology or pulmonary venous flow. No septal defect was observed and there was no evidence of patent ductus arteriosus.
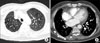
In ventilation-perfusion (V/Q) lung scan analysis, the ventilation scan showed normally distributed Technegas uptake in both lungs but the perfusion scan showed no uptake of Tc-99m macroaggregated albumin (99m-Tc-MAA) in the right lung, indicating a total perfusion defect (Figure 2) and a V/Q mismatch in the right lung.
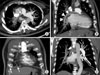
Computed tomography (CT) showed that the right lung was asymmetrically smaller than the left lung. Mild emphysema was observed in both lungs (Figure 3A). Pulmonary vascularity was decreased in right lung (Figure 3B). The diameter of the right pulmonary artery (PA) was small, showing a streaky appearance (Figure 4A). Enhancement of the right PA was subtle, but the right PA was traced to the peripheral portion of the lung (Figure 4B). Vascular hypertrophy was noted in the right intercostal, internal mammary, phrenic and bronchial arteries (Figure 4C, D). A systemic-to-pulmonary shunt through right pleural thickening was suspected.
Because there was a discrepancy between the V/Q scan and the CT image, we performed MRI analysis to evaluate the patient's vascular structures and blood flow in detail.
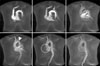
MRI showed hypoplasia of the right PA and dilatation of the left PA. The right ventricle was dilated and the right ventricular wall was hypertrophic. On time-resolved dynamic MR angiography, opacification of the right PA was markedly delayed compared with that of the left PA. The contrast to the right PA was filled after systemic circulation via the aortic arch (Figure 5). Quantitative measurement of blood flow using velocity-encoded cine MR confirmed retrograde flow from the right to the left PA (Figure 6). The peak velocity of main PA flow was approximately 200 mL/s, whereas the peak velocity of right PA flow was 6.0 mL/s (Figure 6A). Moreover, the time-to-velocity curve was reversed in the right PA due to the reversal of flow in this artery (Figure 6B). This retrograde flow in the right PA may be derived from the systemic circulation via hypertrophied systemic arteries surrounding the right hemithorax.
The patient was diagnosed with isolated right PA hypoplasia and severe pulmonary hypertension. He was started on sildenafil citrate first, and then digoxin, furosemide and spinolactone for pulmonary hypertension and right heart failure. After 1 year, he had recurrent hemoptysis and underwent several bronchial and intercostal artery embolizations. During his third admission due to hemoptysis, he died of right heart failure after intercostal artery embolization.
Discussion
To our knowledge, this is the first case report of retrograde pulmonary arterial flow determined by MRI in an older patient with isolated right pulmonary artery hypoplasia. The collaterals are joined to the right PA and flow to the left PA. Systemic collateral flow of an affected lung has been documented in many adults with unilateral pulmonary artery agenesis2,6,7. However, there has been no documentation to date of retrograde flow of the involved artery in UPAH or UPAA by MRI, although its existence has been suggested8.
Blood flow analysis using MRI had a key role in diagnosis. This noninvasive method showed good agreement with the results of angiography and may replace invasive angiography in diagnosing congenital abnormalities of the great vessels. To date, however, despite studies showing aberrant morphology, the blood flow of the involved PA had not been observed4,5,9.
The retrograde flow accounted for the V/Q scan showing a total perfusion defect, although chest CT analysis showed that the right PA could be traced to the peripheral portion of the lung without a cut off. The lung perfusion scan is based on the principle that particles larger than red blood cells are trapped in the first capillary bed encountered after intravenous injection. We infused 99m-Tc MAA into the arm vein of our patient, but it failed to enter the right side despite the pulmonary vasculature visible in the CT scan due to retrograde flow. V/Q scans have been used to differentiate UPAA or UPAH from other similar conditions10,11. The absence of pulmonary artery flow to one lung, accompanied by bilaterally homogeneous ventilation, limits the diagnosis to three main possibilities: pulmonary artery agenesis, thrombotic occlusion and proximal pulmonary artery branch stenosis10,11. Our findings suggest another possibility, retrograde blood flow.
It is unclear whether UPAH is congenital or acquired7,8. Certain clinical characteristics may be useful in differentiating between true congenital absence and hypoplasia of one pulmonary artery accompanied by severe attenuation encountered in chronic lung disease (acquired UPAH)7. Patients with acquired UPAH or unilateral hyperlucent syndrome have lungs with chronic bronchiectasis or other destructive processes and are grossly diseased with markedly impaired ventilation and mediastinal swing during inspiration. Patients with congenital UPAH, however, show normal ventilation in the affected lung, and no or minimal mediastinal swing. In addition, patients with congenital UPAH or UPAA show more abundant systemic collateral vessels throughout the affected lung than do patients with acquired UPAH. Finally, arterial blood gas levels are relatively normal in patients with acquired unilateral lung disease provided the contralateral lung is capable of compensating, whereas those with congenital UPAH or UPAA show hypoxemia due to V/Q mismatch.
We concluded that UPAH in our patient was likely congenital for four reasons. First, an inhalation scan in our patient showed that both lungs were evenly aerated with normal ventilation, and no mediastinal swing was observed on the inspiratory phase of chest MRI.
Second, he had no previous history of severe pulmonary infections and no respiratory symptoms during childhood. Most patients with acquired disease have had severe and recurrent respiratory infections since childhood, resulting in parenchymal destruction8,12. The lung parenchyma was preserved in our patient, except for right pleural thickening with mild fibrosis due to previous tuberculous (Tb) pleurisy and emphysema. Although he had been diagnosed with Tb pleurisy at the age of 57 years, the onset of dyspnea was 2 years earlier. Moreover, despite anti-Tb medications, his symptoms were persistent with no improvement. Also his Tb pleurisy was not sufficiently serious to cause acquired pulmonary artery hypoplasia.
The third reason that UPAH in our patient was likely congenital was our finding, that systemic collateral vessels were distributed throughout the entire area of his right lung. If pulmonary arteries fail to develop, the branches expected to become bronchial arteries thrive unchallenged. Therefore, in congenital disease, there is overall development of the collateral vessels, whereas, in acquired disease, the collaterals consist of one or two enlarged bronchial arteries and small branches7. In our patient, the collaterals were spread throughout the lung, without localized distribution of enlarged bronchial arteries.
Finally, arterial blood gas analysis in our patient showed hypoxemia and increased hemoglobin levels, reflecting chronic hypoxemia. Although our patient had abundant systemic collateral flow to the involved lung, with protected parenchyma and replacement of PA blood for gas exchange, he had hypoxemia owing to a V/Q mismatch. As he became older, his hypoxemia was gradually aggravated.
The patient's UPAH remained unrecognized for a considerable amount of time. As collaterals fed his right lung and his underlying lung diseases of emphysema and sequelae of Tb masked the symptoms and signs such as small hilum, small lung volume, and decreased pulmonary vasculature. The oldest patient previously reported with congenital or acquired, isolated UPAH or UPAA was 66 years old1,13. Therefore, our patient is the oldest to be reported to date.
The terminologies related to UPAH have been used interchangeably as agenesis, aplasia, complete absence, and/or stenosis. Angiographic evidence was used to diagnose UPAA by confirming the absence of the PA. However, failure to fill the PA with contrast does not always indicate absence of the PA. At thoracotomy or on postmortem examination, a patent, albeit small, PA has been observed even when there was no filling of the PA on previous angiography. This might be caused by an increased bronchial arterial flow14. The retrograde flow might run from the bronchial artery to the contralateral PA via the hypoplastic PA. Therefore, we believe that a portion of the UPAA diagnosed by angiography before the CT invention may be UPAH with retrograde flow. The possibility of retrograde flow in acquired diseases was suggested by Raymond and Forsee8 using the dye injection method. We therefore suggest that patients with congenital UPAH have more retrograde flow than those with acquired UPAH as congenital UPAH related to mal-development of aortic arches or pulmonary plexus accompanies more abundant systemic collateral vessels, including bronchial, intercostal, subclavian or subdiaphragmatic arteries15, than those seen in acquired UPAH.
In conclusion, we have described a patient with isolated congenital UPAH and massive collateral systemic flow to the involved lung, which remained minimal and therefore misdiagnosed as emphysema with sequelae of tuberculous pleurisy for a long time. Retrograde flow of the involved pulmonary artery in our patient, a 68-year-old man with isolated right PA hyperplasia, was documented by MRI flow analysis rather than by traditional invasive methods such as pulmonary angiography or retrograde aortogram. Chest MRI and flow analyses were formed due to a discrepancy between uptake in the lung perfusion scan, showing a total perfusion defect in the right lung, and the contrast-enhanced chest CT, showing the presence of the right PA tracing to the peripheral lung parenchyma. Chest MRI with flow analysis is useful in identifying retrograde flow of isolated UPAH with unilateral perfusion defect in a lung perfusion scan and the presence of the involved PA tracing to peripheral lung. Chest MRI may replace more invasive methods in the diagnosis of UPAH or UPAA because it can show cardiac anomalies, mediastinal swing, and systemic collateral flow. Together with an inhalation and perfusion scan, chest MRI with flow analysis may be useful not only to demonstrate retrograde flow when there is a mismatch between perfusion scan and contrast-enhanced chest CT, but also to differentiate between congenital and acquired UPAH.


 XML Download
XML Download