

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The pathogenesis of acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) is a coordinated series of inflammatory process and proinflammatory cytokines play a key role to provoke acute inflammatory pulmonary edema1. Among the proinflammatory cytokines which are responsible for the eliciting acute inflammatory lung injury, interleukin-1 (IL-1) has long been recognized as a causative cytokine to induce ALI by neutrophilic oxidative stress and activation of phospholipase A2 (PLA2)2.
In IL-1-induced ALI in experimental animals, neutrophilic accumulation in the lung and subsequent oxidative stress-induced lung injury are prominent and the activated PLA2 by IL-1 generates proinflammatory eicosanoids3. The oxidative stress in IL-1-induced ALI has been considered as the results of the activation of NADPH oxidase in neutrophils by proinflammatory lipid molecules produced by activated PLA24. As was pointed out, PLA2 is an effector of proinflammatory cytokines and the role of PLA2 in the pathogenesis of ALI is evident in conjuction with neutrophilic respiratory burst5.
Because NADPH oxidase is essential for the initiation of free radical formation from neutrophils6, investigators have tried to ameliorate lung injury by inhibition of NADPH oxidase with apocynin with some successful results. They insisted that apocynin (4-hydroxy-3-methoxyacetophenone) was effective to decrease lung injury through the suppression of oxidative stress by phagocytes7,8. But opposite result was reported also9 and the effect of apocynin on ALI is still controversial.
For the activation of NADPH oxidase in phagocytes, the activation of PLA2, especially cytosolic PLA2 appears indispensable10 and one of the metabolite of cPLA2 activation, arachidonic acid is not only involved in the activation of NADPH oxidase but also takes part in the inflammatory process irrespective of the activation of NADPH oxidase in phagocytes11. For instance, cPLA2-α regulates inflammation by production of platelet-activating factor without NADPH oxidase activity12.
Since IL-1-induced ALI comprises all of the conditions of oxidative stress by NADPH oxidase and activation of PLA2, to test the effect of apocynin on IL-1-induced ALI seems to be worthwhile.
Therefore, in the present study, the effect of apocynin was probed in IL-1-induced ALI associated with oxidative stress and the activation of cPLA2 in neutrophils.
Materials and Methods
1. Animals and reagents
Male Sprague-Dawley rats (300 to 350 g, pathogen free) were purchased from Sasco Korea. Not otherwise mentioned, all experimental reagents were purchased from Sigma-Aldrich Company. (St. Louis, MO, USA). Recombinant human interleukin-1α (IL-1, endotoxin level 1< EU/mg) was purchased from R&D systems (Mineapolis, MN, USA), frozen in aliquots, and thawed daily before use. 125I-labeled bovine serum albumin (125I-BSA) was obtained from ICN Radiochemicals (Irvine, CA, USA). Goat anti-human cPLA2 polyclonal antibody was purchased from Santa Cruz Biochem (Santa Cruz, CA, USA).
2. Induction of ALI
Animals were fed ad libitum till 24 hours before experiments. ALI was induced as described previously13. Fifty nanogram of IL-1 was dissolved in 1.0 mL of normal saline and this solution was insufflated into the trachea. The NADPH oxidase inhibitor apocynin (8 mg/kg)4 was administered through the femoral vein just after instillation of IL-1. To sham treated rats, 1.0 mL of normal saline was instilled into the trachea.
3. Bronchoalveolar lavage (BAL) and count of neutrophils
Lung lavage was performed by cannulating the trachea and instilling 8.0 mL of normal saline with a syringe. Approximately 6.0 mL of lavage fluid was recovered from each rat. After the recovered lung lavage was centrifuged (1,000 g; 10 minutes), the supernatant was collected and stored at -20℃ for measurement of protein. The sedimented cellular pellet was resuspended in 1.0 mL of distilled water and 1.0 mL of Hank's balanced salt solution (HBSS) for 2 seconds to lyse erythrocytes, then centrifuged again. The supernatant was discarded and the pellet was resuspended in 1.0 mL of normal saline. Using 200µL of suspended solution, cytospin slides were prepared and Wright stained for differential cell count. Total leukocyte counts were performed with a hemocytometer and the fraction of neutrophils was calculated by determining the percentage of neutrophils.
4. Measurement of BAL protein
BAL protein was determined as described previously by Brown et al14. using supernatant of BAL. Bicinchoninic acid was used as chromogenic substrate.
5. Measurement of lung leak:
Four and half hours after IL-1 instillation, 125I-BSA (1.0µCi) was injected through jugular vein. One mL of blood was withdrawn 30 minutes later from the right atrium and then assayed for radioactivity. Subsequently, lungs were perfused blood free with normal saline and removed. Lung leak index was calculated as the ratio of radioactivity in the right lung to radioactivity in 1.0 mL of blood.
6. Lung myeloperoxidase (MPO) assay
To assess the accumulation of neutrophils in the lung, lung MPO was determined as previously described by Goldblum et al15. with modification. MPO activity was measured using o-dianisidine as the chromogenic substrate and H2O2 to initiate the reaction. Light absorbance at 460 nM was proportional to MPO activity.
7. Determination of NADPH oxidase assay
Cytochrome-C reduction assay; Neutrophils were isolated as described previously according to Botha et al16. Briefly, the mixture of 15 mL of pentaspan and 0.1 mL of heparin (1,000 units) were mixed with 30 mL of fresh human blood before standing for 40 minutes at room temperature. Then the plasma layered on 74%, 55% isotonic percoll gradient after separation from erythrocytes, centrifuged (1,500 rpm; 20 minutes) and neutrophils were collected from the interface of 74, 55% of percoll gradients. With isolated neutrophils, NADPH oxidase activity was determined by the method of cytochrome-c reduction assay described previously17. Collected suspension of neutrophils was hypotonic lysed for removal of erythrocytes and washed twice with PBS. For cytochrome-c assay, except control samples, phorbol myristate acetate (PMA, 2.5µg/mL) and apocynin (100µM) were added to the neutrophil suspension (2×107 cells) before incubating for 15 minutes at 37℃. Light absorbance at 550 nM was proportional to the reduction of cytochrome-c by free radicals.
8. Determination of γ-glutamyltranspeptidase (GGT) activity in the lung tissue
As an index of oxidative stress in lung tissues, GGT activity was measured as previously described by Meister et al.18 Briefly, 0.2 mL of L-γ-p-nitroanilide (1 mM), 0.2 mL of glycylglycine (20 mM) and 0.4 mL of Tris-Hcl buffer, and 0.2 mL of lung homogenate were added in the tube and incubated at 37℃ for 30 minutes in shaking water bath. The reaction was stopped by adding 0.1 mL of 50 mM of serine borate solution. The release of p-nitroaniline was recorded as the change of absorbance at 410 nM. The specific activity was expressed as µ moles of p-nitraniline released per minute per mg of protein.
9. Histological preparations for examination of alveolar type II cell
Small blocks (1 mm3) of lung tissue were rapidly fixed in the cold 2.5% glutaraldehyde and the small tissue blocks were degassed by the applying of the pressurized air (20 cm H2O) into the sealed vial. The prefixed tissues were rinsed with PBS (pH 7.4) and post fixed 1% OsO4. After dehydrated with graded ethanol-propylene, the tissues were embedded in epoxy-resin. The embedded tissues were polymerized at 37℃ for 12 hours and at 60℃ for 24 hours before ultra thin section. The sections were stained with uranyl acetate and lead citrate before electron microscopy (H600, Hitachi, Japan).
10. Immunohistochemistry of cPLA2 in BAL cells
Cytospinned cells were subjected to staining for fluorescence for cPLA2 in BAL cells. Goat-anti-human-cPLA2 polyclonal antibody (C-20, Santa Cruz, CA, USA) was used as first antibody. The antibody complex was visualized by staining with propidium iodide.
Results
1. Effect of apocynin on the accumulation and the migration of neutrophils
As was shown in Table 1, numbers of neutrophils in BAL were higher in IL-1 treated rats than those of sham treated rats (p<0.001). Apocynin did not decrease the numbers of neutrophils in BAL of IL-1 treated rats. Apocynin did not affect the accumulation of neutrophils by IL-1 either. The MPO activity of the lung was much higher (p<0.001) in IL-1 treated rats compared with that of sham treated rats. But it was not decreased by apocynin.
2. Effect of apocynin on lung leak
Assay of BAL protein and measurement of lung leak index (LLI) revealed the increased protein leak in IL-1 treated rats (p<0.001) compared with those of sham treated rats. Apocynin was not effective to suppress the lung leak in IL-1 treated rats (Table 2).
3. Effect of apocynin on neutrophilic generation of superoxide anion
In phorbol myristate acetate (PMA)-stimulated neutrophils, the amount of reduced cytochrome-c was very much more than that of non-stimulated neutrophils (p<0.001). Apocynin had decreased the generation of superoxide anion from PMA-stimulated neutrophils (p<0.001) (Table 3).
4. Effect of apocynin on the activity of GGT
As an index of oxidative stress, GGT activity was higher in lung tissues of rats given IL-1 than that of sham treated rats (p<0.001). Apocynin was not effective to suppress the increased GGT activity by IL-1 (Table 4).
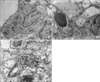
5. Ultrastructural changes of alveolar type II cell
In normal alveolar type II cell, lamellar bodies are well preserved (Figure 1A). By contrast, IL-1 elicited typical morphological changes of oxidative stress in alveolar type II cell such as vacuolization, enlargement of lamellar bodies and disruption of basement membrane (Figure 1B). These pathological changes were not altered by apocynin (Figure 1C).
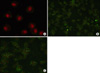
6. Immunohistochemistry of cPLA2 in BAL cells
The BAL cells from the rat given IL-1 showed enhanced expression of cPLA2 in neutrophilic cytoplasms compared with the expression in the cellular cytoplasms of sham treated rats. Apocynin did not alter the increased expression of cPLA2 by IL-1 (Figure 2).
Discussion
IL-1 has been known to be involved in the pathogenesis of ALI by causing neutrophilic oxidative stress19. As an initiator of neutrophilic oxidative stress in cytokine-induced ALI, the significance of NADPH oxidase has long been confirmed20.
In the present study, the inhibition of NADPH oxidase has been tested whether it is effective to reduce lung injury by suppressing oxidative stress in IL-1-induced ALI. Many reports21,22 insisted the beneficial effects of the inhibition of NADPH oxidase by apocynin in various experimental conditions. That is, the inhibition of NADPH oxidase, especially under the condition of neutrophilic accumulation and production of hydrogen peroxide, apocynin was effective to ameliorate the lung injury23.
But in the present study, apocynin did not attenuate ALI induced by IL-1. As was demonstrated in results, IL-1 increased the accumulation of neutrophils in the lung and caused alveolar flooding by the inflammatory process. IL-1 had increased the number of BAL neutrophils and the lung MPO activity. Lung leak and BAL protein had been increased by IL-1 also reflecting the overt acute inflammatory pulmonary edema occurred. Unlike other reports21,22, apocynin was not protective against ALI by IL-1, demonstrated by ineffectiveness of apocynin in reducing lung MPO activity, BAL neutrophils, BAL protein and lung leak index in IL-1 treated rats.
According to the assertion22, apocynin's effects of decreased free radical production has to be potentiated by the MPO and hydrogen peroxide, but in this experiment, apocynin did not show the inhibitory effects on neutrophilic migration and lung leak along with the decreased free radical generation.
Although the inhibition of NADPH oxidase in isolated neutrophils by apocynin decreased the generation of free radicals significantly, the oxidative stress in the lung was not affected by apocynin, which was manifested by the unaltered GGT activity in lung tissues of IL-1 given rats.
Concomitantly, the morphological findings also revealed the ineffectual property of apocynin in diminishing oxidative stress. Alveolar type II cells showed the characteristic changes of oxidative stress24 such as vacuolization and hypertrophy of lamellar bodies in the cytoplasm in IL-1 treated rats. Comparing these changes with the morphology in apocynin treated rats, vacuolization and characteristics of oxidative stress remained.
Clearly, these results reveals that apocynin was not effective to reduce oxidative stress in the lungs of rats given IL-1.
Considering the reports25,26 insisting the protective effects of apocynin on ALI, especially induced by ischemia-reperfusion27, the present data are bewildering and difficult to interpret. But considering the experimental conditions or model for the induction of ALI, there is possibility of other contributing factors causing ALI other than neutrophilic oxidative stress in IL-1-induced ALI in rats. Undoubtedly, IL-1 effected neutrophilic accumulation and oxidative stress as was demonstrated in results. However, the role of neutrophilic oxidative stress might be limited in full blown inflammatory reaction of IL-1-induced ALI.
One of the complexities of pathogenesis in IL-1-induced acute lung injury is derived from the activation of PLA2 by IL-128. By the activation of PLA2, diverse proinflammatory lipid molecules are released from lung tissues and phagocytes. Among these lipid molecules, several lipids are responsible for the activation of NADPH oxidase, but at the same time, some lipid molecules have inflammatory functions irrespective of the function of NADPH oxidase29.
The immunohistochemistry of BAL cells indicated the increased expression of cPLA2 in BAL neutrophils by IL-1. In the BAL cells, cPLA2s were expressed exclusively in the cytoplasms of neutrophils. The increased expression of cPLA2s by IL-1 was not altered by apocynin, and from which it is possible to deduce that the unaffected activation of cPLA2 in neutrophils led to the inflammatory reaction in the lungs of rats given IL-1. According to Brechard and Tschirhart, NADPH oxidase is not inhibited as long as cPLA2 is activated and results in this study are in agreement with this literature30. Another possibility is that free radicals were generated by the activation of cPLA2 without NADPH oxidase. Kim et al. reported that free radicals could be generated in non-phagocytic cells without NADPH oxidase by activated cPLA231.
In addition, some authors insisted that in non-phagocytic cells, apocynin stimulated free radical production instead9. The released arachidonic acid by cPLA2 initiates the activation of NADPH oxidase but at the same time, produces free radicals without NADPH oxidase32.
Collectively, the results from present study suggest that even though apocynin is effective to decrease neutrophilic respiratory burst in vitro, apocynin does not ameliorate IL-1-induced ALI in rats.




 XML Download
XML Download