

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Bacterial infections are related to severe septicemia which is an excessive and uncontrolled innate immune response. In acute lung injury/acute respiratory distress syndrome (ALI/ARDS), which is the most serious consequence of pneumonia and septicemia, inflammatory responses are fulminant and progress in a short time1. The Activation and hyperplasia of cells and remarkable increases in cytokines and chemokines, such as prostaglandins (PGs), leukotrienes, oxygen-free radicals and interleukins, are important in inflammatory responses, and these substances are known to be released mainly from neutrophils and macrophages2. In particular, macrophages play a key role in host defense, wound healing and immunological modulation. Macrophages are also known to regulate mainly innate immunity, phagocytosis and activation of neutrophils as well as acquired immunity including the activation of T lymphocytes and to play a central role in inflammatory immune reactions as antigen-presenting cells3. Macrophages regulate inflammation in ALI/ARDS as well as pulmonary tuberculosis and sarcoidosis. Numerous studies on the role of innate immunity, especially the role of macrophages have been widely conducted in order to elucidate the pathogenesis of ALI/ARDS.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activator protein-1 (AP-1), CREB-binding protein (CBP) and purine-rich box-1 (PU.1) are essential transcription factors after the activation of macrophages through a series of signaling processes4. Macrophages are mainly involved in innate immunity as in sepsis, and PU.1, a transcription factor, regulates the expression of genes that are related to excessive inflammation causing organ failure. However, there have been few studies so far on PU.1. PU.1 is also called spleen focus-forming virus (SFFV) proviral integration site-1 (Spi-1), and it is a member of the EST family of the transcription factors. PU.1 is widely expressed in bone marrow cells and plays a pivotal role in the development of macrophages. PU.1 is activated in macrophages through the activation of a toll-like receptor (TLR)-4 and is involved in various stages of inflammation 4,5. The activation of PU.1 depends on various extracellular stimuli, such as lipopolysaccharide (LPS), GM-CSF and TNF-α, and requires a variety of intracellular signal pathways6. The activation of PU.1 induces the transcription of cyclooxygenase-2 and facilitates the synthesis and secretion of PGs4,5. In particular, PU.1 activates clooxygenase-2 in macrophages. Also, PU.1 is a transcription factor for regulating NADPH oxidase expression in the gp91phox gene7. Lowenstein et al.8 have indicated that PU.1 is involved in the expression of region II of the macrophage nitric oxide (NO) synthase gene.
Proteins in eukaryotic cells attain various physiological functions through the post-translational modification. They are transformed through either binding to phosphate, methyl and acetyl radicals or transient binding to specific proteins. Among the proteins, ubiquitin transiently binding to thousands of different proteins and attains various physiological functions9,10.
Ubiquitins are used classically for nonlysosomal degradation of proteins in proteasome system. Proteasomes has the proteolytic activity for endogenous proteins, which is essential for cellular homeostasis. Protein degradation in this organelle is mediated by polyubiquitination - more than four ubiquitin molecules are chained. But for regulation for transcription factor, monoubiquitin is bound to targeted transcription factor. And this reaction is one of protein-protein interaction, which is bound tightly by covalent bond-isopeptide bond.
So far there is no report for interaction between PU.1 and ubiquitins yet in the macrophages.
Therefore, this study was conducted to examine the ubiquitination of PU.I and its subsequent pathway by LPS stimulation in the macrophages.
Materials and Methods
1. Culture of macrophages
The murine macrophage cell line Raw264.7 (American Type Culture Collection [ATCC], Rockville, MD, USA) was cultured with Dulbecco's Modified Eagle medium (DMEM; GIBCOBRL-Life Technology, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (FBS; GIBCOBRL-Life Technology, Grand Island, NY, USA) and penicillin [100 U/mL)/streptomycin (100 µg/mL)/amphotericin B (GIBCOBRL-Life Technology, Grand Island, NY, USA)in an incubator in a humidified atmosphere of 5% CO2 at 37℃.
2. Primary culture of resident lung, peritoneal and bone marrow macrophages obtained from both BL57/6J wild type and Rac2 null mice
Rac2 null mice were used for primary culture of the macrophages. To separate lung macrophages, a skin incision was made, the trachea was exposed, and a cannula was inserted into the trachea after sacrificing both BL57/6J wild type and Rac2-/- null mice. The trachea was irrigated 10 times with 1 mL of phosphate buffered saline (PBS) and the irrigant was collected in DMEM (10% FBS). To separate peritoneal macrophages, the peritoneal membrane was also irrigated with PBS and the irrigant was collected in DMEM (10% FBS). To separate bone marrow macrophages, the femur was excised, the bone marrow was irrigated with PBS, and the irrigant was then collected in DMEM (10% FBS). After the DMEM solutions were centrifuged, the cell fractions were cultured in a cell incubator after the fractions were well mixed with DMEM (20% FBS).
3. Treatment with reagents
Cells were treated to suppress the proteasome with Escherichia coli LPS (1 µg/mL, Sigma-Aldrich, St. Louis, MO, USA), MG-132 (10 µmol/L; Calbiochem, San Diego, CA, USA) and lactacystin (20 µmol/L; Calbiochem)11 and to suppress the lysosome with ammonium chloride NH4Cl 20 mmol/L; Sigma-Aldrich, St. Louis, MO, USA)12. The cells were pretreated with MG-132, NH4Cl and lactacystin 2 hours before they were treated with LPS.
4. Immunoprecipitation and Western blot analysis: protein-protein interactions (ubiquitination of PU.1)
After the proteins were separated from the cells, immunoprecipitation with antibody and Western blot analysis were performed. The antibody was purchased from Cell Signaling Technology (Beuerly, MA, USA) and experiments were performed according to the manufacturer's instructions.
5. Protein extraction
Cells cultured in a 100-mm dish were washed with ice-cold PBS. M-PER protein extraction solution (Pierce, Rockford, IL, USA) was added to the cells, and the cells were left alone on ice for 1 minute. After that, the cells were scraped and transferred into 1.5-mL tubes. After alternate ultrasonication and centrifugation, lysis buffer was added. The pellet was suspended by pipetting and proteins were extracted by centrifugation.
6. Immunoprecipitation
After 1.5 mL of the protein extract was absorbed to 500 µL of RecA/G-Sepharose beads (Zymed, San Francisco, CA, USA) at 4℃ for 2 hours, the extract was reacted overnight with polyclonal anti-ubiquitin and polyclonal anti-PU.l antibodies (10 µL; Cell Signaling Technology, Grand Island, NY, USA) at 4℃. The protein was precipitated by centrifugation at 12,000 rpm and washed 5 times with 500 µL of PBS. The precipitated protein specimen was denatured by the addition of Laemmeli's sample-loading buffer at 100℃ for 2 minutes and transferred onto SDS-PAGE gel.
7. Western blot analysis
SDS-PAGE was performed at 120 V for 80 minutes. The specimen was transferred into a wet blot chamber, and Western blot assay was performed overnight at using a PVDF membrane at 9 mV. To increase the efficacy of this assay, the PVDF membrane was pretreatd with 6 M guanidine gradient solution (Sigma-Aldrich, St. Louis, MO, USA) containing 20 mM Tris-HCl (pH 7.5), 5 mM β-mercaptoethanol and PMSF. The PVDF membrane was reacted with anti-ubiquitin, anti-PU.1, anti-β-actin and anti-LC3 antibodies (Cell Signaling Technology) overnight. The specimen was washed 3 times with TBS-T solution. After a chemiluminescent (GE healthcare, Piscataway, NJ, MSA) was added, chemiluminescence by horseradish peroxiedase (HRP) was measured using the Chemi-Doc program.
8. NO assay in macrophages
NO was measured with a Griess reagent assay. Briefly, 50 µL each of the standardized reagent and the specimen were placed in a 96-well plate. Alter 50 µL of sulfanilamide solution was added, the specimen was left at the dark at room temperature for 5~10 minutes. Thereafter, 50 µL of NED solution was added, and the specimen was left at the dark at room temperature for 5~10 minutes. NO was measured by spectrophotometry at 520~550 nM.
9. HPLC-mass spectrometry (MS/MS/MS)
PGE2 and PGD2 were measured by the method reported by Cao et al.13 The HPLC system consists of a Shimazu lC-10A pump (Columbia, MD, USA) and a Leap HTS PAL autosampler (Carrboro, NC, USA). PGE2 and PGD2 were measured from a 10-minute linear gradient of 21% to 50% a cetonitrile at a flow rate of 200 µL/min using 10 mM ammonium acetate buffer (pH 8.5) with a Luna phenyl-hexyl analytical column (2×150 mm, 3 µM; Phenomenex, Torranc, CA, USA). Negative ion electrospray tandem mass spectrometry was performed with an API 4000 triple quadruple mass spectrometer (Applied Biosystem, Foster City, CA, USA) at 350℃ at an electrospray of- 4220 V and at a declustering potential of- 55 V. Nitrogen was used at-22 eV as collision gas. During MRM, PGE2 and PGD2 were measured by recording the transition of m/z 351 deprotonated molecules at the most abundant fragment ions of m/z 271. MRM was performed at m/z 355~275 in order to measure internal standards d4-PGE2 and d4-PGD2. The retrieval and analysis of data were performed using Analyst software version 1.2 (Applied Biosystems).
10. Statistical analysis
Data are expressed as mean±standard deviation. Comparisons between the experimental and control groups were made with the paired t test and the Mann-Whitney test. All statistical analyses were performed using SPSS version 16.0 (SPSS Inc, Chicago, IL, USA.) A p-value of <0.05 was considered statistically significant.
Results
1. Western blot analysis of PU.1 according to ubiquitination
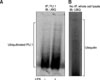
Four hours after the Raw264.7 cells were treated with LPS (1 µg/mL), ubiquitin-PU.1 expression was increased as measured by immunoprecipitation using anti-PU.1 and Western blot assay using anti-ubiquitin (Figure 1A). All the ubiquitinated proteins showed smearing pattern on the PVDF membrane (Figure 1B).
2. Effects of MG-132, lactacytin and NH4Cl on PU.1 expression by LPS treatment
The following experiments were conducted to determine whether PU.1 would be degraded in the proteasome and lysosome after the formation of the ubiquitin-PU.1 complex by LPS stimulation to Raw264.7
cells.
3. Effects of MG-132 and NH4Cl on the expression of ubiquitinated PU.1 by LPS treatment
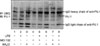
The Raw264.7 cells were pretreated with MG-132 (10 µmol/L) and NH4Cl (20 mmol/L) 2 hours before LPS treatment (1 µg/mL). Two hours after LPS treatment, ubiquitin-PU.1 expression was identified by immunoprecipitation and Western blot assay (Figure 2). When the cells were pretreated with MG-132, ubiquitin-PU.1 expression by LPS treatment was increased (lane 2 vs. lane 5). When the cells were pretreated with NH4Cl, ubiquitin-PU.1 expression by LPS treatment was increased (lane 2 vs. lane 6). When the cells were simultaneously pretreated with MG-132 and NH4Cl, ubiquitin-PU.1 expression was increased (lane 2 vs. lane 8). Ubiquitin-PU.1 expression was stronger in the cells pretreated with either MG-132 or NH4Cl than in those which were not (lane 1 vs. lane 4 and lane 1 vs. lane 3). Ubiquitin-PU.1 expression was stronger in the cells pretreated with MG-132 and NH4Cl than in those that were not (lane 4 vs. lane 5 and lane 3 vs. lane 6).
4. Effects of MG-132 and lactacystin on PU.1 expression by LPS treatment
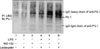
The Raw264.7 cells were pretreated with MG-132 (10 µmol/L) and lactacystin (20 µmol/L) 2 hours before LPS treatment (1 µg/mL). Two hours after LPS treatment, ubiquitin-PU.1 expression was identified by immunoprecipitation and Western blot assay (Figure 3). When the cells were pretreated with MG-132, ubiquitin-PU.1 expression was increased for 24 hours (lane 1 vs. lane 5). When the cells were pretreated with lactacystin, ubiquitin-PU.1 expression was also increased (lane 1 vs. lane 4). Ubiquitin-PU.1 expression was stronger in the cells pretreated with both MG-132 and lactacystin and treated with LPS than in those only pretreated with MG-132 and lactacystin (lane 3 vs. lane 5 and lane 2 vs. lane 4).
5. Early changes in PU.1 expression by LPS treatment (4 hours)
When the Raw264.7 cells were pretreated with MG-132 (10 µmol/L) and treated with LPS (1 µg/mL) for 4 hours, the amount of PU.1 protein was not changed (Figure 4A). In the cells treated in the same manner, PU.1 and ubiquitin were separated by immunoprecipitation using antibodies against PU.1 and ubiquitin, and the amount of the ubiquitin-PU. Complex was measured by Western blot assay using antibodies against PU.1 and ubiquitin. There were no significant differences in the amount of the ubiquitin-PU.1 complex before and after the treatment with LPS, regardless of MG-132 pretreatment (Figure 4B).
6. Late changes in PU.1 expression by LPS treatment (12 and 24 hours)
When the cells were treated with LPS for 12 hours without pretreating them with MG-132/NH4Cl the amount of the ubiquitin-PU.1 complex was not changed (Figure 5, lane 1 vs. lane 2). However, when the cells were pretreated with MG-132 (10 µmol/L) for 2 hours and then treated with LPS, the amount of the ubiquitin-PU.1 complex was increased (lane 3 vs. lane 4). When the cells were pretreated with NH4Cl (20 mmol/L) and then treated with LPS, the amount of ubiquitine-PU.1 was also increased (lane 7 vs. lane 8).
When the cells were treated with LPS for 24 hours without pretreating them with MG-132/NH4Cl, the amount of ubiquitin-PU.1 complex was not changed (Figure 6, lane 1 vs. lane 2). When the cells were pretreated with MG-132 (10 µmol/L for 2 hours and then treated with LPS, the amount of the ubiquitin-PU.1 complex was increased (lane 3 vs. lane 4). When the cells were treated with NH4Cl (20 mmol/L) or both NH4Cl and MG-132, the amount of the ubiquitin-PU.1 complex was also increased (lane 5 vs. lane 8).
7. Changes in NO by LPS treatment after pretreatment with MG-132 and NH4Cl
When the Raw264.7 cells were treated with LPS (1 µg/mL), NO synthesis was markedly increased (Figure 7). When the cells were treated with MG-132 (10 µmol/L) alone, NO synthesis was increased after 24 and 48 hours; however, when the cells were pretreated with MG-132 and then treated with LPS, NO synthesis was not induced as the similar to control group(Figure 7A). When the Raw264.7 cells were treated with NH4Cl (20 mmol/L) alone, NO synthesis was increased after 24 and 48 hours. When the cells were pretreated with NH4Cl and then treated with LPS, NO synthesis was also increased but was lesser than that by LPS treatment alone (Figure 7B).
8. Changes in PGE2 expression by LPS treatment according to the activity of NADPH oxidase
When the alveolar and peritoneal macrophages from the wild type BL57/6J mice were treated with LPS (1 µg/mL), PGE2 synthesis was increased. However, the PGE2 was not detected in Rac2-/- alveolar macrophages. In the peritoneal macrophages, the increment of PGE2 was attenuated in Rac2-/- mice in comparison to in the wild type (Figure 8).
9. Changes in expression of PGE2 and PGD2 in bone marrow derived macrophages (BMDMs) by LPS treatment according to lactacystin pretreatment
When the BMDMs from wild type BL57/6J mice were treated with LPS (1 µg/mL), PGE2 was increased over time; remarkably increased in 24 hours. This increment was augmented when the cells were treated with lactacystin (20 µmol/L) (Figure 9A). In case of PGD2, it was also increased by LPS treatment but this increment was suppressed by lactacystin pretrement, which showed contrast to PGE2 change (Figure 9B).
Discussion
This study demonstrated that ubiquitin-PU.1 expression was increased by LPS treatment in Raw264.7 cells and could be also increased by pretreatment with inhibitors of the proteasome and lysosome, suggesting that the ubiquitin-PU.1 complex is mainly degraded by the proteasomes and lysosomes. This effect occurred in 12 and 24 hours after LPS treatment. We investigated changes in activities of NO synthase and cyclooxygenase-2 due to the accumulation of ubiquitinated proteins. MG-132 is reversible inhibitior of proteasome. Lactacystin is a irreversible inhibitor of proteasome. NH4Cl is inhibitior of lysososme. When these molecules are treated, increased amounts of ubiquitins is expected and ubiquitinated PU.1 is sorted to the proteasomes and/or lysosmes with the subsequent proteolysis. In the macrophages obtained from Rac2 null mice with decreased activity of NADPH oxidase, the proteasome and lysosome were pretreated with their inhibitors. NO expression was changed, and PGE2 was decreased more in the macrophages from Rac2 null mice. PGE2 was increased when the cells were pretreated with the proteasome inhibitor lactacystin, and then treated with LPS. Based on this result, it is inferred that the accumulation of ubiquitinated proteins may play an important role in inflammatory reactions by altering the activities of NO synthase, NADPH oxidase and cyclooxygenase-2.
Macrophages are present in various regions in the human body. They play a central role in innate immunity and recognize external antigens. PU.1, a transcription factor that is expressed at the early stage of development of hematopoietic cells, belongs to the ETS family and acts as a transcription factor specific for monocytes and macrophages14. Approximately 80% of PU.1 is present inside the nucleus, and the remaining 20% is present outside the nucleus. However, the mechanisms of intracellular migration, expression and elimination have not yet been understood. Studies on signaling pathways for PU.1 activation are still primitive. A previous study has reported that the signaling pathway for PU.1 is similar to or related to that for NF-κB such as NIK molecules. When autoantibody against GM-CSF, a stimulant to PU.1, is produced, the resultant abnormalities of PU.1 induce pulmonary alveolar proteinosis, which suggests that PU.1 may be related to physiological functions of macrophages, such as absorption of pulmonary surfactants and maintenance of homeostasis15. PU.1 activates T lymphocytes16. Considering that in the absence of PU.1, leukemia frequently develops and the prognosis of acute infections becomes poor17, it seems likely that PU.1 may be an important transcription factor. Numerous studies on cyclooxygenase-2 have recently been conducted. It has been demonstrated that PU.1 activation induces transcription of cyclooxygenase-2, subsequently promoting synthesis and secretion of PGs4.
The endosome-lysosome pathway and ubiquitin-proteasome pathway are mainly involved in intracellular proteolysis18. The lysosome degrades membrane proteins or extracellular proteins through endocytosis, and monoubiquitin regulates internalization and degradation of membrane proteins through the endosome-lysosome pathway19. The ubiquitin-proteasome pathway, is the main proteolysis process through which intracellular regulatory proteins, abnormal cytoplasm or nuclear proteins are removed. Polyubiquitination is related to this pathway. PU.1 has a PEST domain which is known to correlate with proteolysis by ubiquitin20. In our study, when macrophages were pretreated with MG-132 and lactacystin as inhibitors of the proteasome and NH4Cl as an inhibitor of the lysosome 2 hours before LPS treatment, ubiquitine-PU.1 expression was increased.
Since ubiquitin was first described in 1975, numerous studies have been carried out. Ubiquitin is a protein molecule that covalently binds to target proteins and degrade proteins. The ubiquitin-conjugation pathway involves sequential and step-wise enzymes, and E1 ubiquitin-activating enzyme, E2-conjugating enzyme and E3 ubiquitin ligase form conjngating bonds9. After the polyubiquitination of the target protein, the product moves to the proteasome and degrades proteins. After monoubiquitination, the product involves protein degradation in the lysosome. Previous studies have reported that ubiquitin plays a part in the regulation of the activity of transcription factors, transformation of histone, repair of DNA, phagocytosis of membrane proteins and endocytosis9.
Abnormalities in the ubiquitin system may cause malignant tumors, mental disorders such as Angelman syndrome, degenerative neurological diseases such as Parkinson disease, Huntington disease or Alzheimer disease and type 2 diabetes mellitus.
The ubiquitine-proteasome pathway regulates various transcription factors, such as p53, NF-κB and CBP, which are degraded and lose their activities within the proteasome after being separated from ubiquitin21. The main function of ubiquitin is to regulate amounts of transcription factors in a proteasome-dependent manner. In addition, ubiquitin transforms cofactors, acts as a coregulator, regulates RNA polymerase II in proteasome-independent manner, determines the binding sites of transcription factors within a chromosome and regulates the activities of transcription factors22. Joo et al.23 have shown that the ubiquitin-histone complex relaxes chromosomes and enables transcription factors to bind to the relaxed chromosomes. Gammoh et al.24 have demonstrated that the Mdm2 ubiquitin ligase degrades p53 by directly binding to p53 and induces carcinogenesis by suppressing apoptosis. When inflammation occurs in various conditions suppressed by IKK, IκB is degraded, whereas NF-κB is activated, migrates to the intracellular space and binds to chromosomes through the phosphorylation of IKK and ubiquitination, subsequently expressing genes for inflammation22. Based on these results, it is thought that ubiquitin plays a crucial role in intracellular inflammatory reactions. Bachmaier et al.25 have documented that the acute inflammatory response inderlying lung injury is aggravated with resultant in creased mortality in E3 ubiquitin ligase Cblb knockout mice due to the aggravation of lung injury, suggesting a close relationship between ubiquitin and ALI/ARDS. It is presumed that in a very rapid metabolism as in ALI/ARDS, production of a new transcription factor and its destruction is extremely inefficacious. Therefore, production, transformation, degradation and reuse of a protein are essential to adaptation and maintenance of life phenomena. Based on these results, it is believed that direct protein-protein interactions between transcription factors and ubiquitin are implicated in the regulation of rapid metabolism of proteins.
In this study, when the macrophage cell line, lung macrophages, peritoneal macrophages and bone marrow macrophages were treated with LPS proteins, protein production was changed through the activation of inflammatory enzymes such as cyclooxygenase, NO synthase and NADPH oxidase after pretreatment with an inhibitor of the proteasome or the lysosome. Thus, it is inferred that transcription factors related to inflammation may differ in the degree of ubiquitination. Among them, PU.1 is a transcription factor that regulates expression of cyclooxygenase4, NO synphase8 and NADPH oxidase7 in macrophages. Further studies on regulation of expressions of these enzymes by ubiquitin-PU.1 are warranted. In our study, expressions of PGE2 and PGD2 were changed after treatment with an inhibitor of the proteasome. This may support the result of previous studies that PGE2 promotes inflammation in ALI/ARDS, whereas PGD2 suppresses inflammation26,27. More work is needed to determine how the ubiquitine-proteasome pathway regulates these 2 inflammatory mediators.
Regulation of transcription factors by ubiquitin is a new research field. There have been few studies on clinical implications of PU.1 and regulatory mechanisms of PU.1 in macrophages. Therefore, such studies are thought to be important in elucidating the mechanisms of diseases and developing therapeutic agents for ALI/ARDS. More research into the activation of PU.1 and the role of ubiquitin is warranted in clinical entities such as tuberculosis, sarcoidosis and Langerhans cell histocytosis in which macrophages play a central role in their pathogenesis. It has recently been proposed that ubiquitin-like proteins, including small-ubiquitin-like molecule (SUMO), NEDD and FAT, act like ubiquitin in various cells28,29. Future research should determine whether such actions occur in macrophages.
Several limitations of this study should be addressed. First, for visualization of ubiquitination of PU.1, this study showed only sandwitch exam using anti-PU.1 and anti-ubiquitin antibodies in immunoprecipitation and Western blotting. So it is not excluded that another proteins can be associated with the interaction between PU.1 and ubiquitins. For this discrimination, further experiments such as yeast two-hybrid system, should be needed. Second, for the determination of the transcriptional activation of PU.1, binding to DNA should be also addressed with the method such as chromatin immunoprecipitation. Third, the subsequent pathways after ubiquitination of PU.1 should be conconfirmed. Whether PU.1 ubiquitination is the initiating or terminating step in transcriptional co-activation must be discriminated. PU.1 acts as a transcriptional coactivator and ubiquitination acts a kind of posttranslational modification or protein degradation. Thus, ubiquitination of PU.1 has the meaning of complex feedback mechanism for initiation of genetic expression and termination of genetic expression or proteolysis. Fourth, the clinical meaning of ubiquitination of PU.1 is not sought out yet. Fifth, the remarkable heterogeneity of the macrophages makes it difficult to define the above problems. Sixth, further study is needed about how the relation of upstream and downstream relations amongst TLR, Rac2 (NADPHoxidase), PU.1, COX-2, PGE2, PGD2, NOS, NO. Finally, the determination about how much proportions of ubiquitinated PU.1 is divided into the each of lysosomal, proteasomal and non-proteolytic pathways. Nevertheless, this study is the starting point for overall trial for PU.1 activity in the macrophages, which will enlighten the improvement of clinical outcome for severe inflammatory reaction in the multiple vital organs including the lungs, and the blood streams.
In conclusion, PU.1 may be ubiquitinated with LPS treatment in the macrophages. Further study for determination of mode of ubiquitination and the discrete destiny of ubiquitinated PU.1 according to time sequence should be defined.






 XML Download
XML Download