

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Acute lung injury (ALI), which is called as acute respiratory distress syndrome (ARDS) in its most severe clinical manifestation, affects around 150,000 patients per year in the U.S, with recent mortality rates being over 30 percent1,2. At present, there is no effective treatment for ALI except the low tidal volume ventilation, which is rather a way of reducing artificially induced ventilation-associated lung injury (VLI) than an active, specific way of treatment. ALI is characterized by neutrophil accumulation in the lungs, interstitial edema, disruption of epithelial integrity, and leakage of protein into the alveolar space3-6. Infection, associated with endotoxemia, and blood loss are frequent predisposing factors to the development of ALI. In experimental settings, endotoxemia or hemorrhage produces ALI7. Neutrophils, which play a central role in the ALI, produce proinflammatory mediators, including cytokines such as tumor necrosis factor α (TNF-α) and macrophage inflammatory peptide-2 (MIP-2) and demonstrate increased activation of transcriptional regulatory factors including cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) and nuclear factor kappa B (NF-κB)8-10.
Heparin presents a dazzling array of properties. In lung disease, it is used as an anticoagulant for thromboembolism, but its polyanionic nature confers a wide variety of other actions not related to anticoagulation11. Heparin is a potent antiinflammatoryagent that inhibits neutrophil-derived elastase12, complement activation13, tumor necrosis factor-induced lung edema14, L- and P-selectins15, leukocyte rolling16 and neutrophil-induced injury of pulmonary alveolar epithelium17. However, therapeutic potential of heparin as an antiinflammatory treatment in lung injury is limited by its inherent anticoagulant activity. Attachment of heparin to antithrombin III and some other proteins is critically dependent on binding energies conferred by specific saccharide sequences or charged side groups, and anticoagulant activity can be removed from heparin by partial chemical desulfation18. However, removal of sulfates may have variable effects on other heparin-related activities, which appear to be related to simple charge neutralization of cationic proteins by the anionic polysaccharide11,14. This change often results in the loss of important pharmacological activities. Lyophilization of porcine mucosal heparin under extreme alkaline conditions (pH>13) produces a nonanticoagulant heparin (NCH) remarkable for the selective loss of only 2-O and 3-O sulfates, leaving 6-O and N-sulfates intact. Selectively O-desulfated heparin retains potent activity as an inhibitor of neutrpohil protease, elastase and cathepsin G19. It also inhibited translocation of the NF-κB from the cytoplasm to the nucleus in human endothelial cells and attenuated myocardial reperfusion injury20. It is plausible that NCH could show therapeutic effects on inflammatory diseases of lung. However, as far as authors have searched, there has been no report which evaluated the therapeutic effects of NCH on animal models of the acute lung injury.
In the present study, we evaluated the therapeutic effects of selectively O-desulfated heparin on the mouse model of acute lung injury developed by endotoxemia or hemorrhage.
Materials and Methods
Mice. Male BALB/c mice, 8-12 week of age, were purchased from Harlan Sprague Dawley (Indianapolis, IN). The mice were kept on a 12:12-hour, light-dark cycle with free access to food and water. At least five mice were used for each experimental group. All experiments were conducted in accordance with institutional review boardapproved protocols.
Materials. Nonanticoagulant heparin (2-O,3-O desulfated porcine intestinal heparin, NCH) was kindly donated by Dr. Thomas Kennedy (University of North Carolina, Chapel Hill, NC, USA). Escherichia coli 0111:B4 endotoxin (LPS) was purchased from Sigma (St. Louis, MO). Isofluorane was obtained from Abbott Laboratories (Chicago, IL, USA). Bicinchoninic acid (BCA) protein assay reagent was purchased from Pierce (Rockford, IL). All other reagents were purchased from Sigma unless otherwise noted in the text.
Model for Hemorrhage. Either PBS (200 µl) or NCH (6.25 mg/ml, 200 µl, 50 mg/kg) was injected into the tail vein. Just after the injection of either solution, hemorrhage model was induced into the mouse. In brief, mice were anesthetized with inhaled isofluorane. Cardiac puncture was used to remove 30% of the calculated total blood volume (0.27 ml/10g body wt) over 60 seconds into a heparinized syringe. One hour after the induction of hemorrhage, mice were again anesthetized with isofluorane, and the previously removed blood was infused into the retro-orbital venous plexus. The sham procedure involved cardiac puncture under isofluorane anesthesia, without blood removal, followed by the second episode of anesthesia and retroorbital puncture 1 hour later. One hour after the induction of hemorrhage, Mice were anesthetized with inhaled isofluorane and chest was opened and flushed by infusing 10 ml of PBS into beating right ventricle. After then, lungs were removed and stored at -70 ℃ before being used for cytokines and MPO assay (Figure 1).
Model of endotoxemia. Either PBS (200 µl) or NCH (6.25 mg/ml, 200 µl, 50 mg/kg) was injected into the tail vein. Just after the injection of either solution, endotoxemia-induced lung injury was made by injecting LPS (125 µg/ml, 200 µl, 1 mg/kg) into the peritoneum. One hour after the injection of LPS, mice were anesthetized with inhaled isofluorane and chest was opened and flushed by infusing 10 ml of PBS into beating right ventricle. After then, lungs were removed and stored at -70 ℃ before being used for cytokines and MPO assay (Figure 1).
Preparation of lung homogenate for ELISA. Lung tissues were homogenized in ice cold lysis buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 1 mM sodium vanadate, 10 mM sodium pyrophosphate, 10 mM NaF, 300 µM p-nitrophenyl phosphate, 1 mM PMSF, 10 µg/ml leupeptin, 10 µg/ml aprotinin, pH 7.3) containing 1 mM of protease inhibitor cocktail (Sigma, St Louis, MO). Homogenates were centrifuged at 14,000 g for 15 minutes and supernatants were collected. The protein concentration of each sample was assayed using the micro BCA protein assay kit standardized to BSA, according to manufacturer's protocol (Pierce, Rockford, IL).
Cytokine ELISA. Immunoreactive TNF-α, MIP-2 and IL-1β were quantitated using commercially available ELISA kits (R&D Systems, Minneapolis, MN), according to manufacturer's instructions.
Myeloperoxidase assay: MPO activity was assayed as described as follows. Lung tissue was homogenized in 1.0 ml of 50 mM potassium phosphate buffer (pH 6.0) containing 10 mM N-ethylmaleimide for 30 seconds on ice. The homogenate was centrifuged at 12,000 g for 30 minutes at 4 ℃. The proteinous pellet was homogenized once more in ice-cold buffer, and the homogenate was centrifuged once more at 12,000 g for 30 minutes at 4 ℃. The pellet was resuspended and sonicated on ice for 90 seconds in 10 times vol of hexadecyltrimethylammonium bromide buffer (0.5% hexadecyltrimethylammonium in 50 mM potassium phosphate, pH 6.0). Samples were incubated in a water bath (56 ℃) for 2 hours and then centrifuged at 12,000 g for 10 minutes. The supernatant was collected for assay of MPO activity as determined by measuring the H2O2-dependent oxidation of o-DA (3,3'-dimethoxybenzidine dihydrochloride) at 460 nm.
Wet-to-Dry Lung Weight Ratios. Lungs were excised, rinsed briefly in PBS, blotted, then weighed to obtain the "wet" weight. Lungs were dried in an oven at 80℃ for 7 days to obtain the "dry" weight.
Statistical Analysis. Data are expressed as means ± SEM. ANOVA was performed with SPSS Windows 9.0 statistical analysis software, and a difference was accepted as significant if the p value was less than 0.05, as verified by Duncan and Tukey post hoc test.
Results
The expressions of lung cytokines were increased in endotoxemia model
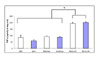
The expression of TNF-α in PBS injected mice (control group) was 83.7±18.4 pg/ml/g of lung protein and that of LPS injected mice was 191.6±10.8 pg/ml/g of lung protein (p<0.05 compared to control group or hemorrhage group). In the while, lung TNF-α level in mice with hemorrhage was 91.1±6.3 pg/ml/g of lung protein (p>0.05 compared to control group) (Figure 2). The lung MIP-2 expressions in the control, hemorrhage, and endotoxemic mice were 187±26, 200±22, and 3000±725 pg/ml/g of lung protein, respectively. The level of lung MIP-2 in the endotoxemic mice was significantly higher than those of control group or hemorrhage group (p<0.05) (Figure 3). The level of IL-1β in endotoxemic mice (6,500±1167 pg/ml/g of lung protein) was significantly elevated compared to those of control group (266±25 pg/ml/g of lung protein) or hemorrhage group (224±24 pg/ml/g of lung protein) (p<0.05) (Figure 4).
NCH treatment had no effects on the expressions of lung cytokines
There was no difference in the expressions of lung cytokines between mice injected by PBS or NCH in control states. In detail, lung TNF-α, MIP-2, and IL-1β levels of PBS- vs NCH-injected mice were 83.7±18.4 vs 57.8±10.4, 187±26 vs 168±33, and 266±25 vs 204±28 pg/ml/g of lung protein, respectively (Figure 2, 3, and 4). Both in hemorrhage and endotoxemic models, NCH injection didn't affect the expressions of three cytokines. In hemorrhage model, lung TNF-α levels in PBS-injected or NCH-injected mice were 91.1±6.3 and 87.5±5.5pg/ml/g of lung protein (p>0.05) (Figure 2). Those of MIP-2 levels were 200±22 and 231±41 pg/ml/g of lung protein (p>0.05) (Figure 3). Lung IL-1β levels were 224±24 and 492±44 pg/ml/g of lung protein (p>0.05) (Figure 4). In endotoxemic model, lung TNF-α levels in PBS-injected or NCH-injected mice were 192±11 and 199±6 pg/ml/g of lung protein (p>0.05) (Figure 2). Those of MIP-2 levels were 3000±725 and 3791±779 pg/ml/g of lung protein (p>0.05) (Figure 3). Lung IL-1β levels were 6500±1167 and 7080±447 pg/ml/g of lung protein (p>0.05) (Figure 4).
NCH treatment did not reduce the recruitment of neutrophils in the lung
The MPO activities in PBS- or NCH-injected control mice were 10.5±2.3 and 5.9±0.7 U/g of lung (p>0.05). Those of PBS- or NCH-injected mice in hemorrhage model were 16.5±3.2 and 21.2±3.0 U/g of lung, which were higher than those of control group but no significant difference between them (p>0.05). In endotoxemic model, MPO activities were increased both in PBS-injected (27.9±6.2 U/g of lung, p<0.05 compared to control group) and NCH-injected mice (32.1±3.6 U/g of lung, p<0.05 compared to control group). However, there was no significant difference in MPO activity between PBS-injected and NCH-injected mice in endotoxemic model (p>0.05) (Figure 5).
Either hemorrhage or endotoxemic model didn't increase in the leakage of lung
Lung leakage was evaluated by wet-to-dry ratio. The wet-to-dry ratios of PBS- or NCH-injected mice in control mice were 4.66±0.06 and 4.68±0.05 (p>0.05). Those of PBS- or NCH-injected mice in hemorrhage mice were 4.73±0.05 and 4.63±0.05 (p>0.05). In endotoxemic mice, they were 4.72±0.03 and 4.77±0.04 (p>0.05) (Figure 6).
Discussion
Sepsis, which is one of the most important causes of acute lung injury, is the leading cause of death in critically ill patients world-widely. In the United States, sepsis develops in 750,000 people annually, and more than 210,000 of them die21. Despite more than 20 years of extensive research, sepsis and systemic inflammatory response syndrome (SIRS) remain the chief causes of death in intensive care units, with mortality rates between 30 to 70%. Further while, according to a recent report, the incidence is rising at rates between 1.5% and 8% annually22. In the while, hemorrhage is often associated with severe acute lung injury23. The ischemia and reperfusion that occur with hemorrhage result in production of reactive oxygen species (ROS) that are thought to cause some of the lung injury that follows hemorrhage24. A common consequence of sepsis or hemorrhage associated with systemic inflammation is multiple organ dysfunction syndrome (MODS)25. Whereas the clinical manifestations of this syndrome are inconsistent, alterations in pulmonary functions are almost always observed26,27. For example, systemic administration of LPS and/or hemorrhagic shock induced an increase in the expression of inflammatory cytokines (TNF-α, IL-6) in serum and in the lung as early as 1 hour after insults7,28,29. Components of the pulmonary dysfunction associated with MODS include widening of the alveolar-arterial PO2 gradient, decreased lung compliance and pulmonary edema.
Most clinical trials targeting blockade of specific inflammatory mediators have not been successful except activated protein C (APC), which has recently been approved for treatment of severe sepsis. In the same sense, transfusion is the sole therapeutic option for the victims of major trauma which doesn't prevent the development of acute lung injury after severe bleeding. In the context of these dire situations in the management of sepsis- and hemorrhage-associated acute lung injury, we tried to evaluate the therapeutic potential of NCH on acute lung injury. Previously in in vitro study, NCH not only decreased the neutrophil-induced injury of pulmonary alveolar epithelium17, but also prevented ischemia-reperfusion injury of the lung30,31. With these actions, heparin might pose an ideal possible treatment for acute lung injury. With these in mind, we evaluated the therapeutic effects of NCH on the two kinds of acute lung injury model (hemorrhage and LPS injection). Both model induced the increase of MPO activity in the lung and in the LPS injection model, the expression of lung cytokines (TNF-α, MIP-2, IL-1β) increased. These results imply that hemorrhage and endotoxemia could result in the development of acute lung injury. Unfortunately, however, NCH failed to reduce the expression of lung cytokines (Figure 2, 3, 4) and neutrophil recruitment (Figure 5). By the way, both models for acute lung injury, endotoxemia and hemorrhage, didn't even increase the leakage of the lung (Figure 6), which was evaluated with wet-to-dry ratio. The validity of this method for the evaluation of lung leakage needs to be tested in the future.
The failure of NCH to reduce the inflammation of lung might be related it's inefficacy in the in viro model of acute lung injury. However, because we tried a fixed dose of NCH (50 mg/kg) at fixed time point (one hour after the induction of acute lung injury), it is still a too hasty conclusion to give off the hope for NCH as a potential agent in the treatment of acute lung injury.
In conclusion, NCH failed to reduce acute lung injury associated with endotoxemia and hemorrhage. However, considering the fact that the history of therapeutic intervention for sepsis has been referred to as 'the graveyard for pharmaceutical companies', this result is not quite surprising. Still the research to find the exact time point of injection and effective dose of NCH needs to be continued.





 XML Download
XML Download