

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Polycystic ovarian syndrome (PCOS) remains one of the leading endocrine disorders encountered by women of reproductive age; however, the etiology and pathogenesis of this syndrome remain an area of continued debate and controversy.
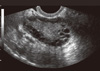
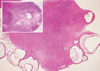
PCOS is considered a syndrome due to heterogeneity in the features of this complex disorder. The Rotterdam criteria were established to confirm diagnosis of PCOS in women who have the presence of at least two of the following symptoms; hyperandrogenism, polycystic ovaries and oligo- and/or anovulation [1]. Hyperandrogenism, an increase in circulating androgen levels such as testosterone, androstenedione, or dehydroepiandrosterone, may lead to a variety of clinical features such as hirsutism, a masculine pattern of facial and body hair growth, presents as an increase in terminal hairs on the lip, chin, chest, back, abdomen, or thigh and acne, inflamed sebaceous glands of the skin [2]. Hyperandrogenemia also causes hair loss (alopecia) on the scalp as a result of sensitivity of the hair follicles to androgen [3]. Polycystic ovaries, the presence of multiple (> 10) cysts in an ovary [4], is caused by the arrest of follicle development at an immature stage (Fig. 1). PCOS is named in reference to this morphological change. As the development of these follicles is arrested well before the point of dominant follicle selection, and therefore positive estrogen feedback to the hypothalamus and pituitary axis is lacking, the LH surge is absent in PCOS patients [5-7]. Consequently, ovulation and menstrual cycles are interrupted (oligovulation and oligoamenorrhea, respectively).
1. Endocrine features
PCOS is widely recognized as an endocrine disorder with numerous metabolic consequences. Unfortunately, the metabolic consequences of PCOS are interwoven with the metabolic consequences of obesity and causal relationships are therefore hard to define (i.e. while PCOS is classified as an endocrine disorder, obesity will affect the endocrine parameters of this disorder). Insulin resistance is one of the metabolic diseases associated with PCOS, and is perhaps the most well documented symptom [8]. The clinical manifestation of insulin resistance at a patient level reflects the complexities described above. Approximately half the women with PCOS are obese [9] and therefore prone to the presentation of insulin resistance [8]. However, insulin resistance is also observed in women with PCOS of a normal body weight [10], together indicating that the hyperinsulinemia associated with this syndrome is not specific to the stressors associated with obesity. The relationship between glucose and insulin is also somewhat patient-specific and affected by age, race, and physical condition. Therefore, insulin resistance/glucose tolerance is a metabolic disease associated with PCOS that can manifest itself in more severe outcomes such as diabetes. Cardiovascular risk is increased in patients with PCOS [11-14] and hypertension [15] is the most easily recognized manifesta tion. This metabolic disorder is also associated with obesity and again, insulin resistance is a known cardiovascular risk factor [11]. Insulin affects myocardial and skeletal energy metabolism and physical activity improves the insulin sensitivity of skeletal muscle [10,16-18]. Downstream effects of exercise-dependent changes in insulin include better blood flow, more efficient glycogen synthase activity and glucose transportation [18]. Similar to the individuality of glucose/insulin tolerances, measures of cardiovascular function such as maximal O2 consumption (VO2max) are also subject to age, physical condition and other diseases with VO2max positively correlated to both testosterone and insulin sensitivity [19].
The relationship between steroids, lipids and glucose metabolism is not easily defined. Interactions between these parameters are known and aberrations of one will affect the other as a metabolic consequence will modulate the endocrine environment. As such, many of the symptoms and features of PCOS play on one another to exacerbate the presentation of the others. The interconnection of the symptoms of PCOS has thus made it a large challenge to pinpoint the causal factor(s) as well as those that are secondary to that initial aberration.
2. Clinical treatments
PCOS is a very complex disorder with heterogeneity of clinical as well as endocrine features. To treat symptoms caused by the hyperandrogenemia of PCOS (hirsutism, acne, seborrhea and hair loss), antiandrogens such as flutamide and spironolactone are routinely administered [20-27]. Most of all, however, treatment of PCOS patients has been centered on inducing ovulation. Due to an inability to naturally ovulate, PCOS patients receive either pharmacological or surgical treatments to induce ovulation together with other assisted reproductive technology (ART) procedures [28-34]. The most commonly used procedures are to increase sensitivity to insulin, induce the gonadotropin surge, or lower circulating testosterone levels. For each of the symptoms, the following treatments are used.
1) Metformin
While the etiology is unclear, it is well established that PCOS and obesity/diabetes are tightly associated. In support of this physiological relationship, weight loss not only improves insulin sensitivity but also often restores ovulatory cycles in some women with PCOS [35-40]. This finding has led to the application of a pharmacological approach for recovering ovulatory cycles in PCOS patients using metformin, an insulin-sensitizing drug [41-45]. Metformin lowers blood sugar levels by decreasing the amount of sugar produced by the liver, increasing the amount of sugar absorbed by muscle cells and decreasing the body's resistance to insulin. Recent studies show that metformin is safe and effective in lowering insulin and improving fertility [46,47].
2) Clomiphene/gonadotropins
For a PCOS patient seeking to become pregnant, the first line of therapy may be to treat her with clomiphene citrate (CC). Clomiphene citrate is a selective estrogen receptor modulator (SERM) that acts as an estrogen receptor antagonist in the hypothalamus [48-51]. When administered, CC prevents the negative feedback effect of estrogen, thus stimulating gonadotropin releasing hormone (GnRH) from the hypothalamus, which in turn increases the secretion of gonadotropins from the pituitary. Therefore, CC is widely used to stimulate folliculogenesis and ovulation in PCOS patients. However, in some women, CC is not successful at inducing ovulation. In these cases, the direct injection gonadotropins can be used to stimulate follicle development and ovulation [52-54].
3) Laparoscopic surgery
In women for whom CC or gonadotropin treatment is unsuccessful, another commonly used approach is ovarian surgery. Although there are several different techniques, they all involve acute ovarian tissue damage. Various types of ovarian surgery have been employed (wedge resection, electrocautery, laser vaporization, multiple ovarian biopsies and others [55-60]. All of these procedures result in a positively altered endocrine profile after surgery. While the current hypothesis is that a small amount of damage to the ovary works to break the cycle of excessive androgen production and abnormal negative feedback, the mechanism behind the reversal of endocrinological dysfunction in PCOS after ovarian surgery remains incompletely understood.
3. Proposed pathogenesis
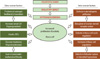
While the initiating factor of PCOS has yet to be determined, we can evaluate the collective pool of known and suspected features of PCOS to better clarify how each symptom relates and what may be driving the symptoms as a whole. In Figure 2, we summarize the known extraovarian and intraovarian factors, and propose causal relationships among them and their effects on inducing hyperandrogenemia and polycystic ovary (arrows). In this particular 'hypothetical' diagram, theca cell hyperplasia, an established cause of PCOS, is proposed as a driving factor for the development of hyperandrogenemia and polycystic ovaries. Of the extraovarian factors, metabolic disorders (abnormal regulation of insulin and/or IGFs) and obesity may stimulate thecal hyperplasia, noting that approximately half of PCOS patients are obese [9]. Hypergonadism caused by dysregulation of LH secretion may also induce thecal hyperplasia, as LH stimulates theca cell proliferation [61-63]. Concurrently, intraovarian factors such as defects in steroidogenesis, regulation of the cell cycle, or ovulatory defects may result in thecal hyperplasia and hyperandrogenemia.
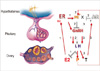
In regard to the mechanism of how hyperandrogenism may lead to ovulatory defect and therefore formation of follicular cyst in the ovary, it has been proposed that elevated testosterone disrupts the regulation of GnRH secretion [64]. In support of this, either GnRH agonists or LH treatment induces ovulation in PCOS patients. A few animal studies have shown that androgen suppresses progesterone receptor (PR) expression in the hypothalamus [65-67]. As progesterone down-regulates GnRH secretion by activating PR activity in the hypothalamus [68-70], androgen-mediated suppression of PR expression may ameliorate GnRH secretion, and therefore LH secretion (Fig. 3). This altered release of gonadotropins will lead to defects in follicle development, the development may arrest outright leading to failure to ovulate (anovulation) or be reduced leading to irregular ovulation (oligo-ovulation). The arrested follicle may thus form a cystic structure, leading to the polycystic ovarian phenotype and loss of fertility in PCOS patients.
Insulin resistance and obesity themselves are also known to negatively affect the secretion of pituitary hormones, and therefore follicular development [71-73]. It is not known whether altered steroidogenesis, pituitary hormone regulation or the defects in follicular development lead to an alteration of metabolism such as insulin resistance and obesity.
4. Animal models for the study of PCOS
To identify the complex nature of this disease, suitable animal models are needed. Researchers during the past three decades have identified different animal models that mimic many of the features of PCOS in women (these are summarized in Table 1). These models have afforded valuable information into complex nature of PCOS. Currently, although a genetic component to PCOS has been identified, a specific "PCOS gene" has not, and therefore a specifically targeted gene deletion for PCOS as an animal model is not available. Therefore, the majority of PCOS models that are available to date rely upon external chemical treatments with steroids, steroid precursors or steroid receptor antagonist to achieve the pathology. Many of these models do not produce consistent results or cease to produce PCOS like symptoms once the treatment stops. Recently, Hill et al. [74] noted that their model for cardiovascular disease, the hypothalamic pro-opiomelanocortin (POMC) specific leptin-insulin double receptor knockout mouse exhibited some similar features to PCOS. Our recently developed theca specific Esr1 specific knockout mouse model reproduces the clinical pathologic features of PCOS in 100% of animals.
1) Prenatal androgen
Prenatal androgen (PNA) treatment in sheep and monkeys results in multiple metabolic and reproductive abnormalities. In monkeys, daily subcutaneous injections of 15 mg of testosterone propionate for 40-80 days gestation are needed to induce the syndrome. In ewes, an injection of 100 mg of testosterone propionate twice a week for 60 days between days 30 and 90 of the 147-day pregnancy result in the ovarian abnormalities. In both models, the abnormalities mirror the symptoms found in women with PCOS. These models produce a long lasting effect in the female offspring mimicking many similar features of PCOS in humans. However, ewes and monkeys incur a large financial commitment for a long gestational period. However, it is noted that although PA monkeys exhibit hyperandrogenemia, the increases are not as extreme as in PCOS women 0.3-0.4 ng/mL (~50-100% elevation above normal) [76,90]; PCOS women, 0.5-0.7 ng/mL (~70-200% elevation above normal) [91-94] and that although anovulation observed in PNA monkeys, its prevalence is also significantly less than that of PCOS women (PA monkeys: ~40%; PCOS women ~90%) [90].
2) Pre-pubertal androgen
This model exploits the association of elevated androgen levels during puberty and PCOS. Immature rats (approximately 21 days old) are treated for 7-35 days with ~100 µg/day testosterone propionate or dihydrotestosterone. Similar to the PNA animal models, pre-pubertal androgen (PPA) animal models of PCOS utilize a unique window where administration of exogenous androgens results in permanent damage to the ovarian tissue and recapitulated the hallmark symptoms of PCOS in an animal model. PPA model shows many similar features to PCOS in women with the exception of the hallmark increase in basal LH levels [80,81]. This model is reliant on artificial hyperandrogenemia and therefore does not help identify abnormalities upstream of hyperandrogenemia.
3) Letrozole
Letrozole is an oral non-steroidal aromatase inhibitor. Inhibition of aromatase prevents the conversion of androgens to estrogens and therefore this model has similar features to the prepubertal androgen treatment [80,82]. Immature rats (approximately 21 days old) are treated for 7-35 days with 400 µg/day letrozole. Similarly to pre-pubertal androgen, this model is also reliant on artificial hyperandrogenemia and does not help identify abnormalities upstream of hyperandrogenemia.
4) Dehydroepiandrosterone (DHEA)
Immature rats or mice (approximately 21-22 days old) are treated with daily s.c. DHEA injections (rats; 6 mg/100 g body weight, mice 6 mg/kg body weight) for 15-20 days. This dose of DHEA is sufficient to induce a hyperandrogenized state similar to that in PCOS women. This model is also reliant on artificial hyperandrogenemia and does not help identify abnormalities upstream of hyperandrogenemia.
5) RU486
Mature cycling female rats are treated daily with RU486 (20 mg/kg body weight) for more than 8 days starting on the day of estrus. These animals exhibit increased basal LH, polycystic ovaries, ovulation blockade and metabolic defects [87]. However, this model is reversible and symptoms decrease upon cessation of the antiprogestin treatment.
6) Estradiol
The immune system is now a well-recognized component of reproductive biology. Immune cells are involved in all aspects of normal reproductive function including ovulation, corpus luteal formation, uterine receptiveness and maintenance of pregnancy. The recently developed PCOS model developed by Chapman and colleagues [88] now provides evidence that aberrant immune function may play a role in the pathogenesis of PCOS and that PCOS may result from as autoimmune disease. This model exploits a 2-3 day window of neonatal period (5-7 days of age) when estradiol administration (20 µg/day) disrupts thymic maturation. As a consequence of this, increased vascular permeability in the thymus allows autoreactive T-cells to escape into the circulation. The ultimate effect of these "escapees" is a damage to tissues throughout the body including the ovaries. This model offers an abrupt change in the direction of PCOS research. The major deficiencies in this model are a lack of hyperandrogenemia and the fact that the loss of regulatory T-cell function would not only impact the ovary, but multiple other tissues resulting in pathologies not associated with PCOS. It will be interesting to follow the progress of this novel concept of autoimmune responses resulting in PCOS in women and if, PCOS women have deficiencies in regulatory T-cells.
7) Hypothalamic pro-opiomelanocortin (POMC) neuron specific leptin and insulin receptor KO
Hill and her colleagues were interested in insulin resistance and the development of type II diabetes when they developed their Pomc-Cre, Leprflox/flox IRflox/flox mice, effectively removing both leptin and insulin receptors specifically from the POMC [74]. However, together with the anticipated glucose intolerance and insulin deficiency these mice suffer from hyperandrogenemia and polycystic ovaries. These two pathological conditions secure its eligibility as a model for the study of human PCOS.
5. A novel hyperandrogenic animal model for the study of PCOS
The unraveling of a biological pathway for PCOS and assigning etiologies to this syndrome requires the development of a novel animal model that consistently present PCOS phenotypes. We therefore launched a project to develop a mouse model of PCOS that is genetically modified and thus stably displays PCOS phenotypes. In doing so, we utilized the underlying concept of a vicious cycle [95-97], where the abnormalities are reinforced. As a way to initiate this cycle, we targeted the ovarian steroidogenic pathway to induce a preference to androgen synthesis and therefore create hyperandrogenemia.
The ovary is comprised of follicles at different stages of development. During follicular growth, the oocyte becomes surrounded by the granulosa cells, a basement membrane forms, and layers of theca cells develop, surrounding the follicle. The two cell layers act cooperatively in the production of androgens and estrogens. Theca cells produce androgens that traverse the basement membrane to the granulosa cells, where aromatase converts the androgens to estrogens. The estrogens in turn negatively feedback to the theca cells causing a decrease of androgen production. This negative feedback action of estrogen is mediated by estrogen receptor 1 (Esr1; ERα) in the theca cells, the prime site of Esr1 expression in the ovary. Upon activation by the binding of estrogen, Esr1 suppresses the expression of Cyp17, a critical enzyme that catalyzes a rate-limiting step in androgen production (Fig. 4), resulting in the decrease of androgen production. Therefore, deleting Esr1 gene would be a logical choice for achieving sustained high levels of androgens because loss of Esr1 will exclude negative regulation of Cyp17 gene expression by estrogen. This will ultimately result in increased production of androgens. The deletion of Esr1 gene has to be, however, limited to theca cells because Esr1 is expressed in so many different tissues/organs including all of the tissues of the hypothalamic-pituitary-ovary-uterus axis [98,99]. Otherwise, the Esr1 deletion in non-theca cells would cause other defects, making it difficult to isolate the Esr1 deletion effect in the theca cells.
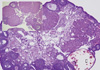
This goal was achieved by selectively knocking out Esr1in the steroidogenic theca cells while keeping the gene intact elsewhere. In principle, this approach would result in the increased expression of Cyp17 gene that encodes a late-limiting enzyme (17apha-hydroxylase) in androgen synthesis pathway, because deletion of Esr1 will relieve Cyp17 transcription from the well-established suppression of this gene by Esr1. We achieved this aim by first generating a mouse line that expresses Cre recombinase under the regulation of the Cyp17 promoter (Cyp17iCre) [100] and then breeding the mice successively with floxed Esr1 (Esr1flox) mice [89]. The offspring with the genotype Esr1flox/flox Cyp17iCre, as was expected, expresses higher level of Cyp17 mRNA expression in the ovary and maintains higher serum testosterone levels (1.5-2 folds over wild type littermates) (ref). The Esr1flox/flox Cyp17iCre mice display other phenotypic symptoms of PCOS patients: (i) irregular estrous cycles, (ii) an age-dependent decrease in ovulatory capacity and fertility defect and (iii) arrest of follicular development at the early antral stage (Figs. 5, 6). This novel animal model generated from the proof-of-principal experiment should bring a new opportunity for the study of PCOS as well as for the study of hyperandrogenemia itself.
6. Future direction
In contrast to the seriousness of the PCOS in women's health, the pathogenesis of this disease is poorly understood. In recent years, efforts by multiple laboratories have led to the development of novel animal models for this complicated disorder. In the next few years, use of these animal models and those to be newly developed will undoubtedly accelerate our understanding on PCOS pathogenesis, and possibly lead to the development of new therapeutic protocols for treating and preventing PCOS in women.


 XML Download
XML Download