

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypoparathyroidism, a disorder of parathyroid hormone (PTH) absence or deficiency, causes hypocalcemia and hyperphosphatemia (1, 2). Postoperative hypoparathyroidism is the most common etiology of hypoparathyroidism (3). Other causes are autoimmune destruction of the parathyroid glands and congenital disorder, such as DiGeorge syndrome (4, 5).
Isolated hypoparathyroidism (IH) is defined as hypoparathyroidism related to an isolated endocrinopathy. IH is a rare disease and its molecular and clinical properties are known to be heterogeneous. In order to overcome the rarity and complexity of IH, several studies have been conducted as a multi-center survey (6, 7). Clinically, patients with IH have shown a broad spectrum of features, from severe symptomatic hypocalcemia in early infancy to asymptomatic, incidental finding in adulthood. Biochemically, IH presents with a wide range of serum calcium and PTH levels. Molecular genetic studies have reported on identification of several genetic mutations, including the prepro-PTH, calcium-sensing receptor (CASR), and glial cell missing B (GCMB) in patients with hypoparathyroidism, however, in most idiopathic cases of hypoparathyroidism, the genetic defect remains unknown (8-11).
Exome sequencing is useful for analysis of selective coding mutations of the human genome for discovery of novel genes for either rare or common disorders. Recently, use of exome sequencing has led to successful discovery of multiple causative gene mutations of genetic diseases (12).
In this study, we recruited 23 patients with hypocalcemia and low serum levels of PTH from eight adult endocrine units in Korea (the Korean Hypopara Registry Study). We present mutational analysis, which can be attributed to the genetic basis of hypoparathyroidism by adopting a whole exome sequencing approach in Korean patients with IH and demonstrate the phenotypic features.
MATERIALS AND METHODS
Subjects
Since 2010, 23 subjects have been included in the Korean Hypopara Registry Study. The inclusion criteria were the presence of hypocalcemia, an inappropriately normal or low level of serum intact PTH, and normal renal function. Patients with post-surgical hypoparathyroidism or other endocrine diseases causing PTH defects were excluded. Blood samples were collected from subjects.
Serum levels of calcium, phosphate, creatinine, and intact PTH were evaluated in all subjects. Ionized calcium, albumin, magnesium, vitamin D metabolites (25-hydroxyvitamin D [25 (OH)D], or 1,25-dihydroxyvitamin D [1,25(OH)2D]) and renal calcium excretion per day were determined in selected subjects.
Whole exome sequencing
Whole exome sequencing was performed using genomic DNA of the affected cases (patients No. 1 and No. 2 in Table 1) and the younger brother of patient No.1 as a negative control at the Theragen BiO Institute (Suwon, Korea). The qualified genomic DNA sample was randomly fragmented using Covaris and the size of the library fragments was mainly distributed between 150 to 200 bp. Adapters were then ligated to both ends of the resulting fragments. The adapter-ligated templates were purified using Agencourt AMPure SPRI beads and fragments with an insert size of approximately 250 bp were excised. Extracted DNA was amplified using ligation-mediated PCR (LM-PCR), purified, and hybridized to the SureSelect Biotinylated RNA Library (BAITS) for enrichment. Hybridized fragments were bound to strepavidin beads, whereas non-hybridized fragments were washed out after 24 hr. Captured LM-PCR products were subjected to an Agilent 2100 Bioanalyzer for estimation of the magnitude of enrichment. Each captured library was then loaded onto a Hiseq2000 platform, and high-throughput sequencing was performed for each captured library in order to ensure that each sample met the desired average sequencing depth. Raw image files were processed by HCS1.4.8 for base-calling with default parameters and the sequences of each individual were generated as 90 bp pair-end reads.
Raw data filtering
Prior to performance of data analysis, removal of dirty raw reads, which only harbor the sequence of the adapter, is essential. The raw data filtering steps were; 1) removal of reads with the adapter, 2) removal of low quality reads (the percentage of low quality bases of quality value≤5 is more than the read). We confirmed a dirty reads rate of 6.6% and a clean reads rate of 93.4%. The mean read depth for the exome sequence was 49x, with 98% of the exome covering clean reads.
Bioinformatics analysis
To improve raw alignment BAMs for single nucleotide variation (SNV) and InDel calling, we performed depth 5-200× and HARD_TO_VALIDATE: MQ0≥4 & (MQ0/[1.0*DP])>0.1) with StandBiasFilter: SB≥-1.0 of condition using GATK. In the next step, patient sample variants were filtered against variants of the control sample and dbSNP data with variants for the InHouse database.
Genetic analysis
Genomic DNA was extracted from peripheral blood from all patients using a standard method. All coding exons and intron-exon junctions for the prepro-PTH (NM_000315.2, NP_000306.1), CASR (NM_000388.3, NP_000379.2), and GCMB (NM_004752.3, NP_004743.1) genes were amplified and sequenced. PCR was performed using 5 mM MgCl2, 200 µM deoxyribonucleotides, 0.5 µM of each primer, 1 unit of Taq polymerase, and 100 ng of genomic DNA as a template. The sequences of primers are available upon request. The PCR products were electrophoresed through polyacrylamide gels and read in both directions using an ABI 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA).
RESULTS
Clinical and biochemical features
We describe baseline clinical and biochemical characteristics of 23 patients (12 males and 11 females) with IH; six subjects of familial type from three families and 17 sporadic cases (Table 1). Three families with IH showed an association with the GCMB mutation (C106R) and CASR mutation (D410E and P221L), respectively. The mean age at onset was 34.6±21.2 yr (range of onset age, 0-67 yr), and three patients had hypocalcemia in early infancy. All patients had a mean calcium level of 6.1±1.1 mg/dL (range 4.4-7.8 mg/dL; Table 1), with a mean phosphate concentration of 5.7±1.4 mg/dL (range 3.2-8.5 mg/dL). For the 22 patients with available data, serum levels of intact PTH were below 8.0 pg/mL in 17 cases or inappropriately normal despite the presence of hypocalcemia (n=5). Vitamin D metabolites were available for 18 patients; mean 1,25(OH)2D level, 38.5± 23.6 pg/mL (n=12) and mean 25(OH)D level, 21.6±11.7 ng/mL (n=16), respectively.
Whole exome sequencing
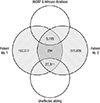
We focused on a family in which a proband and her affected offspring were diagnosed with autosomal dominant hypoparathyroidism (patients No. 1 and No. 2 in Table 1), and performed whole exome sequencing on these two individuals and the proband's sibling, an unaffected individual. A summary of the filtration strategy and numbers of resulting variants is shown in Fig. 1. We obtained 216,260 variants from patient No. 1 and 409,709 variants from patient No. 2, respectively (Fig. 1). We selected the intersection of two sets (patient No. 1 and patient No. 2) and obtained 33,883 variants. Then, we filtered out 27,811 overlapping variants identified from the unaffected individual. The remaining 6,072 variants were then filtered against dbSNP data and the InHouse database, removing all previously reported variants. After filtering, a total of 294 variants (181 single nucleotide variations and 73 insertions with 40 deletions) were identified as being shared by the two affected subjects. All genetic data on the variants are available upon request. Of the 294 variants, we selected 22 candidate genes that reside in coding regions and have lower BLOSUM62 (BLOcks of Amino Acid SUbstitution Matrix) score (http://icb.med.cornell.edu/education/courses/introtobio/BLOSUM62), higher PolyPhen2 score, and lower SIFT score, which might be related to significant amino acid changes and therefore may disease-causing (Table 2). Tables 2 and 3 show final candidate variants related to hypoparathyroidism in patients with familial IH. Among the 22 variants, we noted the C106R mutation in the GCMB gene and confirmed this mutation by direct target gene-sequencing. Functional studies confirmed that GCMB C106R mutant as a loss-of-function mutation could explain hypoparathyroidism in the previous study (11).
Mutational analysis
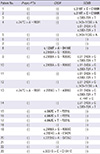
Direct sequencing analysis detected a total of 12 different genetic defects. They are five variants in the GCMB gene, including one functional mutation, six variants in the CASR gene, including two activating mutations and one variant in the prepro-PTH gene. In addition to the C106R mutation of the GCMB gene, four single nucleotide polymorphisms (SNPs) were found in the GCMB gene (c.-44T>C [rs16870746], c.91-242A>G [rs9379881], c.343+163G>A [rs9393726], and c.583-72A>T [rs2076257]) in our cohort (Table 4).
We also found an activating mutation of the CASR gene (c.1230T >A [D410E]) (patient No. 8 in Table 4), which we reported as a novel mutation in the previous study (13). Another mutation of the CASR gene (c.662C>T [P221L]) was detected in one family (patient No. 15, patient No. 16, and patient No. 17 in Table 4); this mutation was previously known as a functional mutation (14). In addition, five SNPs in the CASR gene (c.2205G>A, c.2824G>A [rs76327999], c.2956G>T [rs1801725], c.2968A>G [rs1042636], and c.3031G>C [rs1801726]) were identified from six subjects. Finally, one SNP in the genes encoding prepro-PTH (c.247C>A [rs6256]) was identified in two patients (Table 4).
DISCUSSION
Here we report on a nation-wide study representing genetic and clinical assessment in 23 patients with IH. IH is a heterogeneous group of disorders with various genetic mutations; mainly PTH and CASR genes affecting PTH secretion, and GCMB genes related to parathyroid embryogenesis (15). In our series, IH results from three different mutations which have been described in the literature (P221L in the CASR gene) (14), including two by our group (C106R in the GCMB gene and D410E in the CASR gene) (11, 13). We showed that IH patients possessed several SNPs in the PTH, CASR, or GCMB genes regardless of accompanying functional mutations.
In our search for candidate genes in patients with IH, we made combined use of genomic tools, such as exome sequencing and direct target gene-sequencing. Whole exome sequencing is a new and powerful tool for elucidation of the genetic basis of unsolved Mendelian diseases and could be considered for use as a diagnostic method in congenital diseases (12). However, identification of disease-related variants among a large background of non-pathogenic polymorphism is always challenging (16). In this study, whole exome sequencing revealed that heterozygous c.316T>C (C106R) in the GCMB gene was one of the causal variants responsible for IH. However, other candidate genes identified by whole exome sequencing suggested that each gene might provide insights regarding contributions to hypoparathyroidism. For example, the frequency of HLA-DRB1*01 and DRB1*09 alleles was reported to be higher among patients with idiopathic hypoparathyroidism and the HLA-DRB1 gene showed an association with CASR autoantibody positivity (17). Therefore, testing for HLA-DRB1 mutation might be helpful in the effort to obtain additional information on an autoimmune component of disease among patients with IH in our cohort.
Using a direct sequencing approach, we found four SNPs in the GCMB gene from nine cases. Although mutations in GCMB genes are known to have a role in pathogenesis of hypoparathyroidism, considerable allelic variability for the GCMB gene has been detected in healthy individuals (15); and noncritical single-nucleotide change or non-functional heterozygous mutation in the GCMB gene is easily identified in IH patients (15, 18).
Direct sequencing analysis of the CASR gene in our IH cohort revealed two activating mutations, the D410E mutation and the P221L mutation. In addition, we found four SNPs in the CASR gene. Among these CASR variants, two SNPs (R990G and A986S) were reported as relatively common exonic variants (19). In our study, the R990G allele was detected in four patients (17.4%) and A986S was detected in only one patient. Some studies have reported an association of the A986S polymorphism with serum calcium levels in healthy adults, however, other studies did not report this association (20-23). Genome-wide association scans in European and Indian Asian cohorts indicated an association of A986S with higher serum calcium levels (24). One hypocalcemic patient in our cohort carried A986S as a non-significant polymorphism with allelic alterations in the GCMB and prepro-PTH genes.
We gathered clinical data including biochemical values and levels of vitamin D metabolites. The hypoparathyroidism patients in this study showed similar sex ratio and a broad spectrum of onset age. Most patients in this study showed relatively normal or decreased urine calcium levels. Urinary excretion of calcium is decreased in idiopathic hypoparathyroidism and elevated in patients with gain-of-function mutation of CASR (25). One patient with CASR D410E mutation was hypercalciuric with urinary calcium over 300 mg and other two patients carrying CASR P221L mutation showed normal urine calcium levels. Serum calcium level or vitamin D status was not associated with urinary calcium excretion.
Measurement of 25(OH)D levels is essential to rule out vitamin D deficiency as a cause of hypocalcemia. In classic vitamin D deficiency, PTH hypersecretion compensates for the tendency for the blood calcium to fall (1). Vitamin D insufficiency, serum 25(OH)D level <20 ng/mL, is reported to be relatively common in Korea (26). Vitamin D insufficiency was found in nine patients (56% of measured) in our subjects with low PTH levels. 1,25(OH)2D is regulated by several factors including PTH, calcium and 25(OH)D levels. Serum 1,25(OH)2D is usually reduced in hypoparathyroidism because PTH stimulates the renal 1α-hydroxylase (27). However, synthesis of 1,25(OH)2D can be determined by 25(OH)D concentration in the absence of PTH (27). The level of 1,25(OH)2D in our study varied, probably depending on PTH levels or substrate concentration.
Until now, several studies have attempted to identify the genetic background, prevalence, clinical characteristics and the disease course of IH by conducting a multi-center survey or via a nation-wide registry database (6, 7, 16, 18, 28-30). A German survey recruiting autosomal dominant hypocalcemia patients focused on identification of the CASR gene mutations and clinical phenotypes (7). The Italian Register of Primary Hypoparathyroidism analyzed biochemical data in relation to the Gsα encoding gene, GNAS1 polymorphism (6). The epidemiologic study reported the prevalence of IH was 7.2 per million population in Japan (29). Our study showed the variation of phenotypic expression of IH, which was similar to previous studies. However, we could not identify the prevalence of IH or Korean-specific genetic mutations due to relatively small numbers of cases.
Our study has several limitations. In general, sampling strategies for exome sequencing involve a family-based approach which means sampling from multiple, affected individuals within a pedigree or parent-child trios and an extreme phenotypic approach which means that individuals at both ends of a phenotype distribution are selected for sequencing (16). We performed exome sequencing with relatively small sample numbers. Therefore, further validation of SNP detected from our study should be performed in 23 cases with sufficient additional controls. We only collected baseline clinical data of cohort subjects. Follow-up consequences of IH and details for treatment are needed in order to provide information on the long-term course of disease. Although our study did not report a new cause for PTH-deficient hypoparathyroidsm, genetic assessment showed that patients with IH carried many nonfunctional variants in the GCMB, CASR or PTH genes.
The presence of CASR autoantibodies or anti-parathyroid gland antibodies is known to be involved in the pathogenesis of IH (10, 31). However, the causal unique markers of autoimmune hypoparathyroidism are not yet established; some anti-parathyroid antibodies have the low specificity for parathyroid tissue or anti-CASR antibody positivity varied because of differences in technique (10). Therefore, we focused on the validation of genetic basis in this IH cohort. Further study to investigate the presence of autoantibodies related to hypoparathyroidism might be helpful to identify causes of IH in which the genetic defect remains unrevealed.
The current study, a multi-center collaborative survey conducted for the first time in Korea, demonstrates a variety of phenotypes in patients and detects the genetic mutations responsible. We show that some patients with IH carry significant nucleotide changes in the GCMB, CASR, or prepro-PTH genes. However, in an appreciable number of cases of IH, an advanced diagnostic approach such as whole exome sequencing for identification of unsolved genetic mutations may be needed.



 XML Download
XML Download