

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of liver disease globally [1]. NAFLD is defined as an accumulation of more than 5% lipids (mainly triglycerides) in the liver of people who consume less than 20 g of alcohol per day [2]. When NAFLD progresses, it becomes a form of non-alcoholic steatohepatitis (NASH) with inflammation [34]. Many studies have shown that an increase in NAFLD is closely related to environmental factors, such as diet and physical activity [5]. Excessive dietary sucrose, fructose, and fat are considered to induce NAFLD and its progression [6]. In recent decades, high fructose consumption has become an important factor contributing to NAFLD as well as symptoms of metabolic syndrome, such as insulin resistance, hypertriglyceridemia, and hypertension [7]. Most of the metabolic disorders caused by excessive fructose intake are related to its rapid utilization in the liver, leading to increased hepatic lipogenesis and very-low-density lipoprotein (VLDL) secretion, which finally causes various dyslipidemia [8]. Fructose is principally metabolized by fructokinase to generate fructose-1-phosphate, providing glycerol phosphate and acetyl coenzyme A. The increased production of acetyl-CoA from excessive fructose uptake increases lipogenesis gene expression, resulting in elevated triglyceride (TG) synthesis in liver [9101112]. Recently, a high-fructose diet rodent model has been established and used for studying the pathophysiology of NAFLD and metabolic syndrome [1213].
Disruption in one-carbon metabolism is known as one of the underlying causes contributing to NAFLD pathogenesis and progression [1415]. S-adenosylmethionine (SAM), produced by the adenosylation of methionine, is considered a pluripotent methyl donor providing methyl groups in the synthesis of DNA and RNA, and the methylation of creatine, phospholipids, neurotransmitters, and proteins [16]. SAM is involved in the synthesis of phosphatidylcholine (PC), an important regulator of the progression of liver steatosis to NASH. The synthesis of PC using the phosphatidylethanolamine N-methyltransferase pathway (PEMT), which accounts for 30% of the PC synthesis pathway in the liver, is a SAM methylation-dependent pathway [141718]. Methyl-donor supplementation to obese mice reduced liver TG accumulation and increased the phosphorylation of genes that inhibit fat synthesis [15].
Numerous studies have shown that AMP-activated protein kinase (AMPK) plays an important role in regulating hepatic lipogenesis [1920212223]. High fructose-induced NAFLD characteristically reduced AMPK phosphorylation, resulting in the increased synthesis of lipids and the decreased synthesis of VLDL, leading to lipid accumulation in the liver [2021]. In the rodent model of fatty liver induced by a high-fat diet, supplementation with folic acid increased AMPK phosphorylation level and decreased fatty liver [202223]. Recent studies suggested that dietary methyl-donor or folic acid supplementation can prevent the progression of high-fat diet-induced NAFLD and improve hepatic cholesterol and glucose metabolism by enhancing AMPK signaling [1823]. Fructose overloading not only stimulates lipogenesis but also increases lipogenic gene expression, leading to lipid deposition in the liver. Acute and chronic fructose intake increases intrahepatic de novo lipogenesis. However, the effect of folic acid supplementation on a high-fructose diet animal model has not been yet reported. Thus, the present study was designed to investigate whether folic acid supplementation is effective in improving high-fructose diet-induced lipid metabolism and demonstrate the underlying mechanisms by which folic acid supplementation reduces hepatic steatosis.
Go to : 
MATERIALS AND METHODS
Animals and diets
The animal experiments were approved by the Institutional Animal Care and Use Committee of Hannam University (No. HNU 2017-03). Five-week-old male Sprague-Dawley rats were obtained from Raon Bio Co (Yongin, Korea). After one week of adaptation, the rats were randomly divided into 3 groups (n = 8/group; average weight: 178 g): control (C), high-fructose diet (HF), and high-fructose diet with folic acid (HF+FA). The formulations of the purified diets are shown in Table 1. The present recommendation for a standard rat diet from the National Research Council is 0.68 percent dietary metabolic energy as linoleate (C18:2) [2425]. In this study, lard was used as a source of fat. However, due to the sufficient linoleate content in the diet, there will be no physiological effects due to the fat source (Table 1). The energy composition of the control diet consisted of 64% of carbohydrates from cornstarch and maltodextrin, 20% protein, and 16% fat (4 kcal/g of diet, Research Diets, New Brunswick, NJ, USA). The HF diet contained 64% fructose. The HF+FA diet was made to contain 64% fructose and 40 mg folic acid/kg diet with reference to previous studies [2223]. All animals had free access to food and water. The animals were housed individually in cages in a room with a 12-h light-dark cycle with 50±7% relative humidity for eight weeks. Bodyweight was measured once per week. Food intake was recorded every three days.
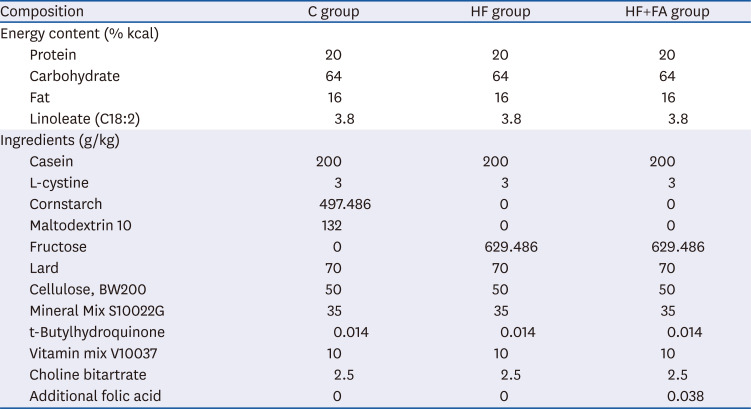
Table 1
Composition of the experimental diets
Vitamin mixture ingredients (g/kg): Vitamin E acetate 15.0, Niacin 3.0, Vitamin B12 2.5, Biotin 2.0, Pantothenic acid 1.6, Vitamin D3 1.0, Vitamin A acetate 0.8, Pyridoxine HCl 0.7, Riboflavin 0.6, Thiamine HCl 0.6, Folic acid 0.2, Phylloquinone 0.08. Mineral mixture ingredients (g/kg): Calcium carbonate 357.00, Potassium phosphate 196.00, Sodium chloride 74.00, Potassium citrate 70.78, Potassium sulfate 46.60, Magnesium Oxide 24.00, Ferric citrate 6.06, Zinc carbonate 1.65, Sodium metasilicate 1.45, Manganese carbonate hydrate 0.63, Copper carbonate 0.30, Chromium potassium sulfate 0.28, Boric acid 0.08, Sodium fluoride 0.06, Nickel (II) carbonate 0.03, Sodium selenite 0.01, Potassium iodate 0.01, Ammonium molybdate tetrahydrate 0.01, Ammonium (meta)vanadate 0.01.
C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid.
![]()
Blood glucose and biochemical analysis
In the last week of the feeding period, the rats were fasted overnight, and then blood was drawn from the tail vein. Fasting blood glucose was measured using a glucometer (CareSens; i-SENS, Inc., Seoul, Korea).
At the end of the feeding period, the rats were fasted overnight (12 h) and anesthetized using carbon dioxide. Blood was collected in ethylenediaminetetraacetic acid (EDTA)-coated vials by cardiac puncture and plasma was collected after centrifugation of the blood at 3,000 ×g for 15 min. The plasma was kept frozen at −70°C until analysis. The plasma levels of TG, total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), glutamate-pyruvate transaminase (GPT), and glutamate-oxaloacetate transaminase (GOT) were determined by using commercial kits (Asan Pharm Co., Ltd., Seoul, Korea).
Liver SAM and S-adenosylhomocysteine (SAH)
SAM and SAH were analyzed by high-performance liquid chromatography (HPLC) equipped with a C18 Betasil column (250 mm × 4.6 mm Ultrasphere 5-µm particle size) (Thermo Fisher Scientific, Runcorn, UK) using a gradient mobile phase. The frozen liver was homogenized with 0.4 M perchloric acid and then centrifuged at 12,000 ×g at 4°C for 30 min. The concentrations of SAM and SAH were quantified from the absorbance of the peak at 254 nm [26].
Hepatic lipid analysis
After collecting the blood, the livers were removed, rinsed with physiological saline solution, and weighed. The liver tissues were divided into small pieces and immediately frozen in liquid nitrogen before being stored at −70°C until analysis. A piece of liver tissue was fixed in 10% formalin for the preparation of paraffin sections. For hepatic lipid analysis, the liver lipids were extracted as described by Folch et al. [27]. Hepatic TG levels were measured by commercial kits (Asan Pharm Co., Ltd., Seoul, Korea).
Hepatic histology
Hepatic lipid accumulation and morphologic changes were visualized in liver cryosections (5 µm). After the livers were embedded in paraffin, the sections were stained with hematoxylin and eosin (H&E). Optical microscopy analysis was performed by using a Leica DMIL LED optical microscope (Leica, Wetzlar, Germany).
Western blot analysis
Liver tissue was homogenized using Tris-HCl buffer (pH 7.5) containing 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% NP-40, and 1% phenylmethylsulfonyl fluoride (PMSF). The tissue extracts were centrifuged at 12,000 ×g at 4°C for 20 min and the protein concentration of the supernatant was determined using a BioRad protein assay according to the manufacturer's protocol (Bio-Rad Laboratories, CA, USA). Equivalent amounts of protein of each sample were separated by 12% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Shanghai, P.R. China). The membranes were then blocked with 5% skim milk and subsequently incubated with the appropriate primary antibodies liver kinase B (LKB1) (Cat No. 3047), phospho-LKB1 (Cat No. 3482), AMPK (Cat No. 2532), phospho-AMPK (Cat No. 2531), acetyl-CoA carboxylase1 (Cat No. 3676), and phospho-acetyl-CoA carboxylase1 (Cat No. 3661) (all from Cell Signaling Technology, Inc., Danvers, MA, USA), and β-actin (Cat No. SC-47778, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The membranes were subsequently reacted with horseradish peroxidase-conjugated secondary antibody (Cat No. 7074S). The immunoreactive bands were visualized by incubation with lumiGLO reagent (Cell Signaling, Beverly, MA, USA) and analyzed using an LAS 4000 chemiluminescent image analyzer (Fuji, Tokyo, Japan).
Statistical analysis
All statistical analyses were performed using SPSS (v 23.0) for Windows (IBM Corp., New York, NY, USA). The data are presented as the mean ± SD of 8 rats per group. Where appropriate, the data were analyzed using one-way analysis of variance, followed by Duncan's multiple range post-hoc test. The minimal level of statistical significance was set at P < 0.05 in all analyses.
Go to :
RESULTS
Bodyweight and liver weight
As presented in Fig. 1, we observed gradual body weight increases in all 3 groups during the experimental period. The body weights were slightly lower in the high-fructose-fed groups (HF and HF+FA) compared to the C group, although there was no significant difference between the 3 groups (Fig. 1). The liver weight and the food intake of the C, HF, and HF+FA groups were not significantly different (Table 2).

 | Fig. 1Effect of folic acid supplementation on the bodyweight of high-fructose-fed rats. Male SD rats were fed C, HF, or HF+FA groups for 8 weeks. The results are presented as the mean ± SD (n = 8). Data were analyzed by ANOVA.C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid; ANOVA, analysis of variance.
|
Table 2
Effects of folic acid supplementation on body and liver weights in high-fructose-fed rats
The data are the mean ± SD (n = 8).
C, control; HF, high-fructose diet, HF+FA, high-fructose diet with folic acid; NS, not significant.
![]()
Blood glucose, plasma lipids, and liver function enzymes
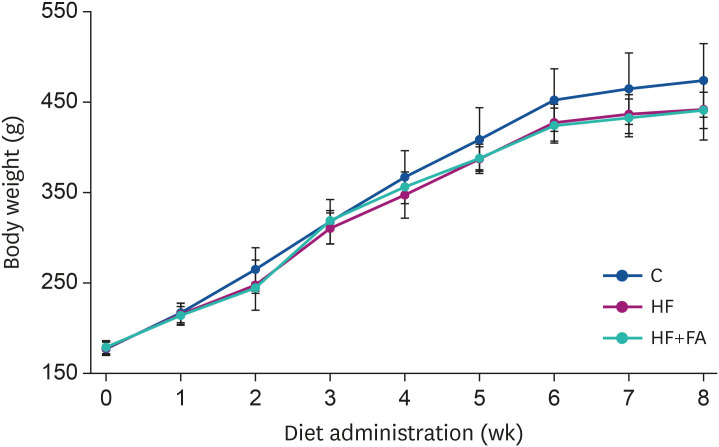
At the end of the study, the fasting blood glucose levels were not significantly different in the HF group compared to the C group (Fig. 2A). However, the fasting blood glucose in the HF+FA group was significantly lower than in the C and HF groups (P < 0.001).
 | Fig. 2Effects of folic acid supplementation on fasting blood glucose, GPT, and GOT in high-fructose-fed rats. Male SD rats were fed C, HF, or HF+FA groups for 8 weeks. Each bar presents the mean ± SD (n = 8).GPT, glutamate-pyruvate transaminase; GOT, glutamate-oxaloacetate transaminase; C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid; ANOVA, analysis of variance.
a-cDifferent letters mean significant difference according to ANOVA, Duncan's test (P < 0.05).
|
The plasma levels of GPT, a marker of liver injury, were significantly increased in the HF group compared to the control (44.3 ± 5.6 vs 18.6 ± 4.7 IU/l, P < 0.000). The plasma GOT levels of the HF group were also increased about 1.6-fold higher compared to the control (95.3 ± 10.1 vs 60.2 ± 6.5 IU/l, P < 0.000) (Fig. 2B and C). Supplementation of the high-fructose diet with folic acid significantly reduced the plasma GPT levels compared to the HF group, but there was no significant effect of folic acid on the plasma GOT levels.
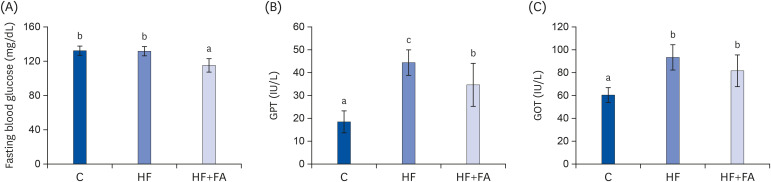
At the end of the diet feeding period, the plasma TG and TC in the control rats were 157.2 ± 49.0 mg/dL and 94.1 ± 7.2 mg/dL, respectively (Fig. 3A and B). Feeding the HF diet greatly elevated the plasma TG (P < 0.05) and slightly elevated the TC (P < 0.000) compared to the C group. Folic acid supplementation reduced the plasma TC (P < 0.05) in rats fed the high-fructose diet. However, the plasma HDL-C concentrations were similar in the control, HF, and HF+FA groups.
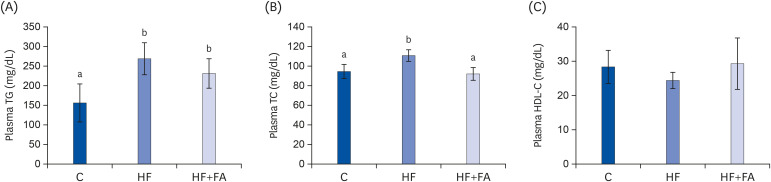 | Fig. 3Effects of folic acid supplementation on plasma lipids in high-fructose-fed rats. (A) Plasma TG (B) Plasma TC (C) HDL-C. Each bar presents the mean ± SD (n = 8).TG, triglyceride; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; ANOVA, analysis of variance; C, control; HF, high-fructose diet; HF+FA: high-fructose diet with folic acid.
a,bDifferent letters mean significant difference according to ANOVA, Duncan's test (P < 0.05).
|
Liver lipid profiles and liver histology
To examine the hepatic lipid contents, we extracted the total lipids from liver tissue and analyzed the total lipids and TG. The hepatic TG level of the HF group was 21.8 ± 5.8 mg/g liver (P < 0.01), about 1.9-fold higher compared to the C group (Fig. 4A). The hepatic TG level of the HF+FA group was significantly lower compared to the HF group (Fig. 4A). The hepatic total lipids of the HF group were 109.0 ± 20.3 mg/g liver (P < 0.01), about 1.5-fold higher than the C group (Fig. 4B). Although supplementation of the high-fructose diet with folic acid significantly reduced the liver TG levels, no difference in liver total lipids was observed in the HF+FA group compared to the HF group (Fig. 4B).
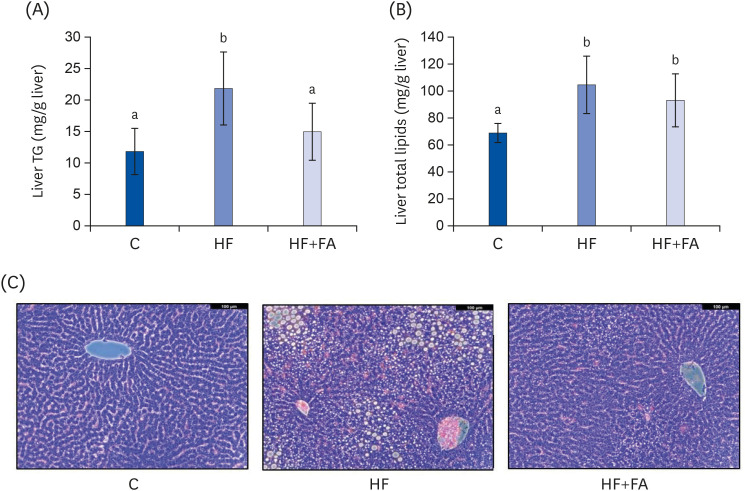 | Fig. 4Effects of folic acid supplementation on liver lipid accumulation in high-fructose-fed rats. (A) Liver TG (B) Liver total lipid content (C) Liver sections stained with H&E. Each bar presents the mean ± SD (n = 8). The images were captured using a Leica DMIL LED optical microscope (Leica, Wetzlar, Germany).TG, triglyceride; H&E, hematoxylin and eosin; ANOVA, analysis of variance; C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid.
a,bDifferent letters mean significant difference according to ANOVA, Duncan's test (P < 0.05).
|
The microscopic examination of H&E staining of liver tissue was consistent with the hepatic lipid biochemical characteristics (Fig. 4C). Exacerbated macrovesicular steatosis with single, large fat droplets and smaller well-defined fat droplets, as well as intense inflammatory infiltrates, were observed in the HF group. Supplementation of a high fructose diet with folic acid significantly reduced the number and size of fat droplets and the inflammatory infiltrates compared to the HF group.
Liver SAM, SAH, and SAM:SAH ratio
After eight weeks of dietary treatment, the liver SAM, SAH, and the SAM:SAH ratio in the HF group were significantly lower compared to the C group (Fig. 5). Supplementation of the high-fructose diet with folic acid significantly increased hepatic SAM levels, but did not change the SAM:SAH ratio compared to rats fed a high-fructose diet.
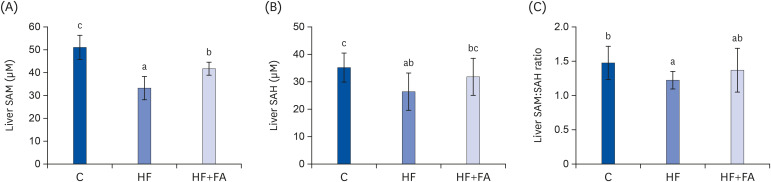 | Fig. 5Effect of folic acid supplementation on liver SAM, SAH, and SAM:SAH ratio in high-fructose-fed rats. (A) Liver SAM, (B) Liver SAH, and (C) Liver SAM:SAH ratio. Each bar presents the mean ± SD (n = 8).SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; ANOVA, analysis of variance; C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid.
a-cDifferent letters mean significant difference according to ANOVA, Duncan's test (P < 0.05).
|
AMPK activation and de novo fat synthesis
To elucidate the mechanism of folic acid supplementation in alleviating the fat accumulation in the liver of high-fructose diet-fed rats, we measured the protein expression levels of the LKB1-AMPK pathway regulation molecules, including LKB1, AMPK, and acetyl coenzyme A carboxylase (ACC) in liver tissues by Western blotting (Fig. 6). It is known that the phosphorylation of LKB1 and AMPK and the dephosphorylation of ACC are important for the activation of the LKB1-AMPK pathway [28].
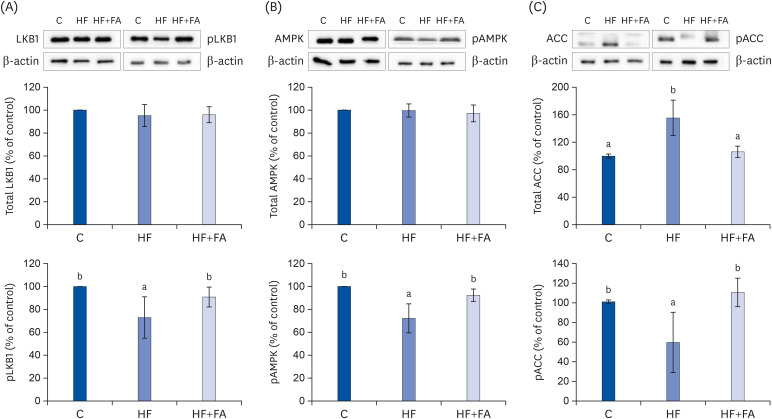 | Fig. 6Effects of folic acid supplementation on hepatic expression of pLKB1 & LKB1 (A), pAMPK & AMPK (B), and pACC & ACC (C) protein in high-fructose-fed rats. Each bar presents the mean ± SD (n = 8).ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; LKB1, liver protein kinase B1; C, control; HF, high-fructose diet; HF+FA, high-fructose diet with folic acid.
a,bDifferent letters mean significant difference according to ANOVA, Duncan's test (P < 0.05).
|
A high-fat diet has been suggested to induce fatty liver by modulating the AMPK pathway in the NAFLD animal model fed a high-fat diet. In this study, feeding a high-fructose diet significantly inhibited AMPK activation compared to the C group. High-fructose diet inhibited the activation of LKB1, a kinase upstream of the AMPK pathway, compared to the C group. The relative expression levels of phospho-LKB1 were restored by folic acid supplementation (Fig. 6A). In addition, folic acid supplementation also restored the relative expression levels of phospho-AMPK (Fig. 6B). Because AMPK activation suppresses fatty acid synthesis by inhibiting ACC, the rate-limiting enzyme in the fatty acid synthesis pathway, we examined the expression levels of ACC in liver tissue. The level of phosphorylated ACC was significantly decreased in the HF group compared to the control. The expression of phospho-ACC was significantly increased in the HF+FA group, which was consistent with the possible suppression of fatty acid synthesis by folic acid supplementation in the rats fed a high-fructose diet.
Go to :
DISCUSSION
Several studies reported that methyl donors attenuated NAFLD induced by animal models fed high-fat diets [202223] However, there are very limited studies regarding the effect of folic acid supplementation on NAFLD induced by a high-fructose diet. In the present study, we aimed to investigate whether folic acid supplementation could prevent fat accumulation in the liver using a high-fructose diet model and identify the underlying mechanism by which folic acid supplementation reduces hepatic steatosis. The high-fructose diet-mediated fatty liver model can be considered a useful model to mimic the pathophysiology of NAFLD in non-obese people compared to a high-fat diet model [7].
The current study showed that the high-fructose diet did not change the liver and body weights, but significantly elevated hepatic TG, plasma TG, and plasma TC levels. Similar to our observation, in a study by Mahesh et al. [29], a high-fructose diet induced fatty liver without significant differences in body or liver weight for 8 weeks. Similarly, Kawasaki et al. [7] reported that, in a study of rats fed a high-fructose, high-fat, or high-sucrose diet for 6 weeks, there was no significant difference in the body weight or liver weight in the high-fructose or high-sucrose-diet groups compared to the control group. In contrast, bodyweight was significantly increased in the high-fat diet group. Thus, a high-fructose diet has been suggested as a model for inducing NAFLD without weight differences from the control group in various studies [30]. These characteristics of the NAFLD model induced by high-fructose may be due to the unique metabolic properties of fructose in the liver.
We observed that high fructose consumption resulted in hepatic fat accumulation without body weight change and an approximately 2-fold increase in liver TG and total lipids compared to the control group (Fig. 4). In addition, folic acid supplementation (40 mg/kg diet) significantly reduced the histologically and biochemically measured liver fat in rats fed a high-fructose diet and, thus, appeared to prevent many pathophysiological abnormalities in NAFLD. Plasma GPT and GOT are representative indicators for the diagnosis of liver function and liver disease. GPT is present only in cytoplasm and most sensitively reflects the degree of necrosis of the liver [31]. GOT is an enzyme in the cells of the heart, liver, muscle, and red blood cells. Chronic fatty liver damages mitochondria and destroys organ cells, which leads to elevated GPT in the plasma. The increased plasma GPT and GOT in the high-fructose group demonstrated that the high-fructose diet induced liver damage and steatosis leading to NAFLD. We observed that folic acid supplementation significantly reduced plasma GPT, indicating that the folic acid supplement used in this study was not at a toxic level and confirming that folic acid supplementation prevented liver damage induced by high-fructose intake.
We also observed that hepatic fat accumulation was induced by high fructose intake in the H&E-stained histological sections. As shown in Fig. 4, folic acid supplementation suppressed the fructose-induced increases in liver fat accumulation. These results demonstrate that folic acid supplementation improved high-fructose-induced liver steatosis and damage.
SAM and SAH are the substrate and product, respectively, of the many cellular methyltransferases and the ratio of SAM and SAH is frequently used as an indicator of cellular methylation capacity [26]. Chronic high fructose intake can stimulate fat synthesis due to abnormal increases in acetyl-CoA in the liver and a decrease in SAM, leading to the impaired secretion of VLDL, which results in fat accumulation in the liver. In addition, the accumulation of fat in the liver was achieved by decreasing the phosphorylation of AMPK in the liver, increasing fat synthesis, and decreasing the beta-oxidation of fat. The beneficial effects of folic acid supplementation may be due to reductions in liver fat accumulation and the recovery of liver SAM levels by folic acid supplementation (Figs. 4 and 5). In the present study, we showed that folic acid supplementation was able to prevent fatty liver when given in conjunction with a high-fructose diet by maintaining SAM levels which is essential for lipid export from the liver. The majority of the metabolic disorders related to fructose is reported to be related to its rapid utilization through the liver, leading to increased hepatic lipogenesis and the secretion of VLDL from the liver, which finally causes various dyslipidemias [81014]. SAM is a universal methyl donor for all methylation reactions, including glycine n-methyltransferase and PEMT, all of which produce SAH, the principal inhibitor of all methylation reactions. The PEMT pathway using SAM can synthesize the choline moiety of PC and phosphatidylethanolamine N-methylation is regulated mainly by hepatic SAM concentrations, which respond to the SAM:SAH ratio [3233]. Because PEMT, the liver-specific SAM-dependent methyltransferase, is essential for lipid export from the liver, low SAM concentrations and SAM:SAH ratio could lead to abnormal lipid accumulation in the hepatocytes. In this study, the SAM and SAH concentrations and the SAM:SAH ratios were decreased in the livers of rats fed excess fructose (Fig. 5), and folic acid supplementation given in conjunction with high fructose increased liver SAM. Therefore, our findings demonstrate that folic acid supplementation may contribute to increasing the synthesis of VLDL in the liver of high-fructose-fed rats and thereby, improving the release of lipids from the liver. Recent studies in the methionine adenosyltransferase (MAT) 1A knockout model showed that NASH was induced when the liver lacked SAM [34]. The biosynthesis of PCs reduced by SAM deficiency is closely related to NASH development, implying the need for methyl donors to prevent non-alcoholic fatty liver. Decreased hepatocellular SAM:SAH ratios could ultimately produce a defect in the secretion of VLDL and contribute to hepatic fat accumulation in rats consuming a high-fructose diet. The ability of the methyl donor to increase this ratio and thus, maintain optimal PEMT activity and restore VLDL secretion, may explain the ability to attenuate fatty liver.
Since AMPK regulates the metabolic pathways involved in energy metabolism, it has been proposed as a potential therapeutic target in metabolic diseases including NAFLD [3536]. LKB1 is known to be activated by phosphorylating AMPKα when intracellular ATP decreases and AMPK increases [37]. LKB1 appears to direct most of the AMPK activation in the tissues examined so far, with the exception of some cells [38394041]. Phosphorylated AMPK is involved in regulating lipid metabolism by catalyzing the phosphorylation of ACC. The phosphorylation of ACC inhibits its enzymatic activity, thereby reducing triglyceride synthesis in the liver [2842].
We performed Western blot analysis to determine the mechanism by which folic acid supplementation improved liver fat accumulation. We found that the high-fructose diet reduced the phosphorylation of AMPK and supplementation with folic acid given in conjunction with the high-fructose diet restored the phosphorylation of AMPK to normal (Fig. 6B). In addition, supplementation with folic acid decreased the total ACC and increased phospho-ACC through increased AMPK phosphorylation. The increase in phospho-AMPK by folic acid supplementation was associated with an increase in phosphorylated (inactivated) ACC, suggesting decreased hepatic fatty acid synthesis with folic acid supplementation. ACC is an important enzyme made in the early stages of lipid synthesis by catalyzing the reaction forming malonyl CoA from acetyl CoA [17]. The converted malonyl CoA is synthesized as saturated fatty acid by fatty acid synthase (FAS). This process proceeds to synthesize TG from long-chain fatty acids [43]. The phosphorylation of ACC by AMPK inactivates ACC, decreasing the rate of lipid synthesis [44].
AMPK regulates lipid synthesis, fatty acid degradation, and fatty acid oxidation through the phosphorylation of key substrates. ACC and fatty acid synthase, the two rate-limiting enzymes in fatty acid synthesis, are regulated by AMPK, an enzyme whose activation is related to energy metabolism regulation in the liver. To examine the consequence of significant increases in AMPK phosphorylation, LKB1, a kinase upstream of the AMPK pathway, was also analyzed by Western blots. The phosphorylation of LKB1 in the folic acid supplemented group was increased compared to the high-fructose group. Overall, our results suggest that folic acid supplementation appears to reduce the increased hepatic de novo lipogenesis induced by a high-fructose diet by activating the LKB1, AMPK, and ACC pathways. In an in vitro study on the mechanism by which folic acid activates LKB1 in cultured HepG2 cells, LKB1 activation by folic acid was found to be mediated by protein kinase A [23].
In conclusion, the current study suggests that folic acid increased the phosphorylation of ACC through the AMPK signaling pathway and increased the level of liver SAM, thereby inhibiting liver lipid synthesis and accumulation, improving the liver steatosis and damage induced by a high-fructose diet. This study was a short-term animal experiment conducted for eight weeks, with the limitation that the high-fructose diet did not cause severe NAFLD during the period.
Go to :
 XML Download
XML Download