

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cholestatic syndromes are inborn or acquired disorders of impaired bile flow, which can be caused by either intrahepatic or extrahepatic disorders [12]. In recent years, the identification of genes responsible for different types of cholestasis has greatly contributed to its diagnosis. ABCB11 on chromosome 2q24 encodes the bile salt export pump (BSEP), and functional impairment of this protein results in intrahepatic cholestasis. Pathogenic mutations of ABCB11 are well known causes of progressive familial intrahepatic cholestasis (PFIC), but are also associated with milder forms including benign recurrent intrahepatic cholestasis (BRIC), intrahepatic cholestasis of pregnancy (ICP), and oral contraceptive-induced cholestasis (CC). Clinical differentiation of BRIC and PFIC may be difficult because both syndromes can share similar clinical and biochemical profiles in terms of acute cholestatic attacks. Often diagnosis in BRIC patients is delayed without any conclusion [3]. A number of patients with BRIC are subjected to invasive investigations, and liver transplantation might also be recommended. Diagnosis can be more challenging when BRIC appears in newborns or in early infants. Only clinical course can distinguish those children at the severe end of the disease spectrum from those at the benign end.
Normal gamma glutamyl transpeptidase (GGT), elevated bile acid concentration, and conjugated bilirubin in serum are common findings of BRIC, PFIC1, and PFIC2. Serum aminotransferases are higher or markedly elevated in PFIC, whereas normal or moderately elevated aminotransferases are usual in BRIC [3]. The most distinctive differentiation of the two syndromes is clinical course after early exclusion of extrahepatic biliary atresia [456]. Patients with PFIC progress to liver cirrhosis and will require liver transplantation later in life, whereas attacks in patients with BRIC last from several weeks to months and resolve spontaneously. Between attacks, patients with BRIC remain healthy with normal biochemical levels for months to years [7]. For early diagnosis of ABCB11 spectrum liver disorders, we performed next generation sequencing (NGS) on patients with severe neonatal intrahepatic cholestasis to identify the underlying etiology. Consequently, we discovered novel mutations in PFIC2 resulting in early life-saving liver transplantation and the prevention of unnecessary liver transplantation for cases of BRIC2 aggravated by concomitant neonatal viral hepatitis.
MATERIALS AND METHODS
Subjects
Subjects included 50 patients presenting neonatal cholestasis who had visited the Department of Pediatrics at the Kyungpook National University Children's Hospital between January 2013 and July 2016 (Table 1). Detailed clinical and family histories were obtained. Evaluations included laboratory examination, liver ultrasonography (USG), diisopropyl iminodiacetic acid (DISIDA) scan, percutaneous cholecystocholangiography (PCC), and liver biopsy. Genetic analysis was performed when a patient was suspected of having a syndromic disease. Two families also provided genetic material for analyses to confirm the diagnosis and were subjected to genetic counseling. Blood samples were collected and genomic DNA was extracted using a QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany). DNA quality and quantity were assessed using a Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA) and a Quant-iT BR assay kit (Q32850; Invitrogen). The institutional review board of Kyungpook National University Hospital approved the protocol, and informed consent forms were obtained for genetic analysis and for utilization of the results for diagnosis and research purposes from the participants or from their legal guardian (IRB no. KNUH 2016-06-011).
Whole exome sequencing
Whole exome sequencing was performed on patients with ABCB11 spectrum liver disorders to analyze multiple candidate genes simultaneously. Targeted exonic regions were captured with the Agilent SureSelect XT Human All Exon v5 kit (Agilent Technologies, Santa Clara, CA, USA). Exome sequencing was performed on an Illumina HiSeq-2000 (Illumina, San Diego, CA, USA) platform with 100-bp paired-end runs at an average mean target depth of 100× coverage. Rare or novel mutations were prioritized.
In silico analysis
The raw sequence data in fastq format were mapped to the reference human genome (UCSC hg 19, NCBI build 37.1) using the Burrows-Wheeler algorithm. Alignments for each sample were converted to binary alignment map (BAM) format, sorted, and indexed, and polymerase chain reaction duplicates were marked and then merged into one BAM file using Samtools. Alignments from the combined BAM file were then locally realigned around insertions/deletions, recalibrated, and variants were called for all samples together using the genome analysis toolkit. Mutations were further evaluated using bioinformatics software programs such as snpEff, SIFT score, polyphen-2 score, phyloP score, phastCons score, 1000 genomes, dbSNP (dbSNP137), and the Korean Personal Genome Project.
Sanger sequencing
Sanger sequencing was performed on the patient and the family members to confirm suspected pathogenic mutations identified from whole exome sequencing and to determine whether other family members without obvious illness had inherited the same mutation. The target site of the variant and the flanking DNA sequences from each family member were amplified with forward and reverse primers. The amplified products were directly sequenced using an automated DNA sequencer (ABI3130; Applied Biosystems, Foster City, CA, USA) using a Big-Dye Terminator Cycle Sequencing kit version 3.1. The primer sequences are available upon request.
RESULTS
Subject characteristics
Fifty patients were divided based on the underlying cause of disease. Idiopathic or viral hepatitis was diagnosed in 17 (34%), metabolic disease in 10 (20%), total parenteral nutrition induced cholestasis in 8 (16%), extrahepatic biliary atresia in 7 (14%), genetic disease in 5 (10%) including PFIC2 and BRIC2, neonatal lupus in 1 (2%), congenital syphilis in 1 (2%), and choledochal cyst in 1 (2%) patient (Table 1).
Idiopathic hepatitis was confirmed as the sporadic form by clinical course, which presumably was undefined viral hepatitis. Of the 10 patients with metabolic disease, six were citrin deficient, two had hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, and two had ornithine transcarbamylase deficiency. Of the five with genetic disease, two had siblings with PFIC2, one had BRIC2, one had Edward syndrome with heart failure, and the other had arthrogryposis-renal dysfunction-cholestasis syndrome.
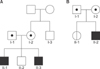
Two families with proven ABCB11 spectrum liver disorders were subjected to genetic analyses (Fig. 1). Biochemical findings in the patients showed markedly elevated levels of aminotransferase with normal GGT levels.
PFIC2
In family A, with Korean ethnicity, genetic testing was performed on the second son (II-2), the third son (II-3), the parents (I-1 and I-2), and the maternal uncle (I-3). There was no access to the first son's (II-1) DNA because he had died 13 years before the study. The first and third sons had progressive cholestasis from the neonatal period. The parents of the affected siblings were non-consanguineous and healthy and there was no family history of liver disease.
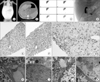
The first son showed persistent jaundice from 7 days after birth and visited a hospital at 2 months of age. Diagnostic evaluations indicated intrahepatic cholestasis with normal GGT levels. As PFIC had not been reported in Korea at the time, genetic testing was not performed, and progressive cholestatic hepatitis did not lead to a specific clinical diagnosis. A liver biopsy was performed in the 1-year-old patient who showed significant pathological features of cholestasis. Liver transplantation was recommended but the parents deferred the decision. At the age of 27 months, the patient was referred to our hospital for urgent management of severe abdominal distension. A plain abdominal X-ray showed a markedly distended abdomen with no air-fluid levels or free gas under the diaphragm (Fig. 2A). Abdominal paracentesis with free drainage was performed and there was hemorrhagic ascitic fluid. Abdomen computed tomography revealed multinodular hepatocellular carcinoma and hemoperitoneum due to the ruptured tumor (Fig. 2B). He expired despite intensive care after moving to another tertiary hospital. The second son did not have any symptoms related to cholestatic liver disease.
The third son displayed similar symptoms and visited our hospital presenting with jaundice from 7 days after birth. Physical examination showed mild hepatomegaly and initial biochemical analysis indicated conjugated hyperbilirubinemia (6.8 mg/dL) and elevated levels of aspartate aminotransferase (AST), 100 U/L; alanine aminotransferase (ALT), 53 U/L; serum bile acid, 124 µmol/L; with normal ranges of ammonia, 40 µmol/L; and GGT, 31 U/L. Liver USG revealed normal hepatic parenchymal echogenicity with a visible common bile duct, and triangular cord sign was not identified. A DISIDA scan, obtained 6 hours after Tc99m injection, did not reveal passage to the small bowel (Fig. 2C), but PCC demonstrated a contrast-filled gall bladder and excreted contrast material in the small bowel loops, which could exclude extrahepatic biliary atresia (Fig. 2D). A liver biopsy was performed at 3 weeks after birth and indicated mild portal inflammation and lobular disarray (Fig. 2E), mild portal and focal periportal fibrosis by Masson's trichrome stain (Fig. 2F), mild bile canalicular and lobular cholestasis with mild ballooning change, focal lobular inflammation, and apoptosis (Fig. 2G). Electron microscopy revealed distended canaliculi with loss of microvilli (Fig. 2H) and filamentous bile, which is a distinctive finding of PFIC2 (Fig. 2I). Cholestatic hepatitis progressed drastically and maximum conjugated bilirubin, AST, and ALT were 11.4 mg/dL, 8,000 U/L, and 3,740 U/L, respectively. After the genetic confirmation of PFIC2, treatment with 4-phenylbutyric acid (4-PBA) was started but showed no effectiveness. The patient underwent liver transplantation from a living donor at the age of 6 months, and is currently recovering and showing normal liver function.
BRIC2
In family B with Korean ethnicity, genetic testing was performed on the parents (I-1 and I-2), daughter (II-1), and son (II-2). The son presented with increasing jaundice since birth. He was born as a full-term baby following a normal pregnancy and delivery. He was the second child of healthy unrelated parents with no family history of liver disease. Upon physical examination, he was normal except for mild hepatomegaly. Initial liver function tests showed conjugated hyperbilirubinemia with a level of 6.2 mg/dL, high alkaline phosphatase (ALP) of 875 U/L, AST of 464 U/L, and ALT of 357 U/L. GGT were within normal limits. Liver USG, a DISIDA scan, and PCC showed unremarkable results and a liver biopsy demonstrated similar findings to that of the PFIC2 patient, distended canaliculi with loss of microvilli (Fig. 2J) and filamentous bile (Fig. 2K).
He was positive for IgM antibodies to herpes simplex simplex virus (HSV), but negative for other viral work ups. Acute cholestatic attack lasted for more than 10 months, and maximum ALP, conjugated bilirubin, AST, and ALT were 884 U/L, 8.2 mg/dL, 1,538 U/L, and 1,141 U/L, respectively. There was the possibility of PFIC2 because the onset was neonatal, the biochemical manifestations were severe, and the acute phase was relatively long. Genetic analysis demonstrated no pathogenic mutation for PFIC2. After 4-PBA was initiated, biochemical improvement occurred dramatically and serum bilirubin and liver enzymes normalized completely within several months. This benign evolution was consistent with a diagnosis of BRIC2.
Identification of mutations in ABCB11 by whole exome sequencing
Whole exome sequencing was performed on the third son of family A and the son of family B, which generated over 50 million reads. Common variants were filtered and the candidate genes associated with inherited cholestatic disease were analyzed. Forty-one variants of ATP8B1, ABCB11, and ABCB4 were identified in the third son of family A. Among those 41, only two novel mutations in ABCB11 (NM_003742.2), c.11C>G (p.Ser4*) and c.1543A>G (p.Asn515Asp), were suspected to be causative mutations. The nonsense and the missense mutations were shown to be conserved in orthologs of mammalian ABCB11, and in silico analyses also demonstrated high probabilities of pathogenicity. These mutations have never been reported in PFIC2 patients or in the general population.
In the son of family B, 13 variants of ATP8B1 and ABCB11 were detected. Among those, ABCB11 c.1331T>C, p.Val444Ala (rs2287622), c.3084A>G, and p.Ala1028Ala (rs497692) had the potential to be causative mutations. The allele frequencies of Val444Ala and Ala1028Ala are 74.5% and 67.2%, respectively [8]. Although both variants are commonly found in the general population, Val444Ala has previously been reported in ICP and drug induced cholestasis with a higher allele frequency than in normal controls [910]. Val444Ala has also been reported in BRIC2 and ICP when present in combination with other ABCB11 or ABCB4 mutations [1112]. Ala1028Ala promotes exon skipping in vitro and is possibly associated with primary biliary cirrhosis [1314].
Validation by Sanger sequencing
The mutations were validated by Sanger sequencing in the patient and family members. In family A, the patient had combined heterozygous mutations of Ser4* and Asn515Asp. The patient inherited Ser4* from the mother and Asn515Asp from the father. The mutations were not identified in the other family members (Fig. 3). In family B, Val444Ala was homozygous in the patient and sister, and heterozygous in the parents. Ala1028Ala was heterozygous in the patient, sister, and father, but homozygous in the mother.
DISCUSSION
PFIC is a group of autosomal recessive disorders that disrupts bile acid secretion and presents with persistent intrahepatic cholestasis leading to early liver failure and death or liver transplantation before adulthood [456]. PFIC is divided into three types based on genetic cause: PFIC1 caused by mutations in ATP8B1; PFIC2, caused by mutations in ABCB11; and PFIC3, caused by mutations in ABCB4. The estimated prevalence of PFIC is between 1/50,000 and 1/100,000 and PFIC2 represents half of all cases. The initial presentation and clinical course of PFIC2 tend to be more severe than those of the other types [5]. Signs and symptoms of cholestasis usually appear in the neonatal period often resulting in end stage liver disease and/or progression to hepatobiliary malignancy within the first years of life. Liver histology of PFIC2 patients is more severe with lobular and portal fibrosis and inflammation than in those with PFIC1, and hepatocellular necrosis and giant cell transformation are more remarkable [7]. Imaging studies show a normal biliary tree, but can reveal biliary stones or a large gallbladder [478]. Extrahepatic features are not present in PFIC2 because ABCB11 is only expressed in hepatocytes [7].
BRIC comprises a group rare autosomal recessive disorders characterized by recurrent episodes of cholestasis without progression to liver failure [4]. Severity and duration of the acute cholestatic attacks vary both clinically and biochemically [5]. BRIC was previously believed to be a milder form of PFIC, but only recently was recognized as a different genetic liver disease [4]. BRIC is divided into two types based on the genetic cause of the condition; BRIC1 is caused by mutations in ATP8B1, whereas BRIC2 is caused by mutations in ABCB11. The usual onset is in childhood and early adolescence, but it can start at any age [6]. Liver biopsies are characterized by intrahepatic cholestasis with preservation of normal liver architecture [6].
ABCB11 gene mutations have been identified in Asian populations including those in in Thailand, India, Japan, Taiwan, and mainland China and is ethnicity-specific [8]. Rapid identification of the etiology is important for prompt management and good prognosis of cholestasis. Of the many conditions that cause neonatal cholestasis, genetically identifiable disorders represent more than 50% of the cases.
In our study, genetic confirmation of ABCB11 spectrum liver disorder led to early liver transplantation in the PFIC2 patient. In addition, the atypically severe BRIC2 patient avoided unnecessary liver transplantation after genetic analysis. Likewise, genetic studies are becoming increasingly important to uncover the underlying etiologies of neonatal cholestasis.
We report two novel pathogenic mutations in PFIC2, and an association between Val444Ala and Ala1028Ala in BRIC2. Val444Ala was reported in BRIC2 in combination with E186G, and this compound heterozygosity of the BSEP gene reduced levels of the BSEP protein due to protein instability or mistargeting [11]. Our BRIC2 patient and his sister had homozygous Val444Ala and heterozygous Ala1028Ala mutations. Ala1028Ala is thought to modify Val444Ala, promoting exon skipping. Ala1028Ala by itself, however, is not pathogenic because the homozygous mother was not a BRIC2 patient. Cholestatic events in patients with BRIC2 are sometimes associated with preceding viral infections [15]. In our BRIC2 patient, HSV infection was a provocative factor for the acute episode. Although the sister had the same genetic basis, she was a 5-year-old at the time of the study and did not have any history of precipitating viral infection. The usual onset of BRIC2 is during adolescence, and thus intensive and regular follow-up for the sister is required.
Clinical experience suggests that drugs traditionally used, such as ursodeoxycholic acid, antihistamines, and cholestyramine, have limited and variable effects on cholestatic attacks. However, rifampicin accelerates the hepatic detoxification and excretion of compounds, and has been shown to be effective for cholestatic attacks in patients with BRIC and PFIC [1617]. Our patients with ABCB11 spectrum liver disorders were treated with 4-PBA instead of rifampicin. As a clinically approved pharmacological chaperone drug, 4-PBA has been shown to target some mislocalized BSEP mutants to the canalicular membrane [1819]. The drug enhances cell surface protein expression for some of the missense mutations found in ATP8B1 and ABCB11 deficiency [2021], and one study showed that bile acids act as pharmacological chaperones of E297G BSEP [22]. As 4-PBA showed complete resolution of acute cholestatic attacks in BRIC2 patients, it could become the first choice of therapy for BRIC2. Whereas our PFIC2 patient with two novel mutations did not show any improvement with 4-PBA treatment, genotypic correlation studies might be needed.
Diagnosing genetically heterogeneous cholestatic disorders is challenging due to the large number of candidate genes [18]. Until a decade ago, a number of cases of infantile cholestasis had been considered idiopathic [19]. However, as genetic testing was developed, more and more patients were retrospectively found to have genetic bases of cholestasis [23]. If whole exome sequencing could have been performed for the first affected son who died in January 2003, he could have survived through an early decision to perform liver transplantation with a confirmed diagnosis. Thus, genetic testing should be performed on all affected patients with PFIC and on their parents [24]. Families with PFIC patients should be offered genetic counseling and prenatal diagnosis [2526]. Rapid advancement of NGS technologies will play a key role in detecting pathogenic variants of inherited cholestatic disease including PFIC. Massive parallel sequencing has enabled the simultaneous screening of huge panels of candidate genes. In conclusion, the diagnostic evaluation of PFIC remains challenging, but NGS represents a promising diagnostic tool for a large number of idiopathic cholestatic disorders. In conclusion, BRIC2 and PFIC2 share similar characteristics in terms of acute cholestatic attacks; a comprehensive genetic analysis will facilitate optimal diagnosis and treatment.


 XML Download
XML Download