

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The discovery of human embryonic stem cells (hESCs) has raised hopes for curing diseases that currently have a dismal prognosis [1]. However, after more than a decade of research, several challenges related to ESC safety, efficacy, and bioethics have not been sufficiently answered. For example, in 2009 the United States Food and Drug Administration (FDA) approved a clinical trial of hESC-derived oligodendrocyte progenitors in spinal cord injury patients, but the trial was subsequently suspended pending further data regarding safety issues (http://www.medicalnewstoday.com/articles/162269.php).
In 2006, it was demonstrated that mouse fibroblasts could be reprogrammed into a pluripotent state similar to that observed in ESCs by the retroviral transduction of the Oct4, Sox2, Klf4 and c-Myc genes [2]. Spurred by this landmark study, human induced pluripotent stem cells (hiPSCs) have been successfully generated from human embryonic, neonatal, or adult fibroblasts [3-5]. hiPSCs have been generated from patients with various diseases [6-10], with several groups reporting disease-specific phenotypes when these cells subsequently differentiate to directed functional cells [9, 11-19].
The recent advances in iPSC technology have made patient- and disease-specific human cells widely available. Patient-specific iPSCs have been derived as sources for drug screening, toxicology, cell replacement therapy as well as generating disease models. For example, for patients with end-stage liver disease, liver transplantation is the only method of treatment [20]. However, the limited availability of donor livers and immunological incompatibilities are major obstacles to liver transplantation. Therefore, alternative methods with the potential to substitute for liver transplantation are required. In recent years, the interest in liver cell therapy has been increasing continuously [21]. From the clinical point of view, transplantation of hepatocytes or hepatocyte-like cells could represent an alternative, either to liver transplantation in acute liver failure or for the correction of genetic disorders resulting in metabolically deficient states. The use of ex vivo adult human hepatocytes is a desirable option for cellular therapies or drug testing. However, these cells have limited proliferation potential, and lose function and viability upon isolation. Although there have been great advances in liver stem cell biology [22-24], hepatic stem cells are infrequent within tissue, making their isolation and expansion unfavorable for large-scale applications [25]. Attempts to immortalize hepatocytes by introducing telomerase constructs and viral transfections also suffer the shortcomings of phenotypic changes, poor liver function, and karyotypic abnormalities [6, 26]. Recently there has been a focus on deriving human hepatocytes from other sources, in particular hESCs and hiPSCs [27-30]. This holds great promise as an unlimited hepatocyte source.
Advances of iPSC Generation Methods
The original method of iPSC induction used a retrovirus vector for transgene expression [2]. Most patient-specific iPSCs have been established with retroviral vectors. However, the retrovirally derived iPSCs have numerous transgene integrations in the genome, and the integration may result in leaky expression, which could disturb the endogenous transcription factor network and lead to the failure of differentiation. Another important problem of transgene integration is the risk of tumorigenesis after transplantation. In particular, the reprogramming factor c-Myc is a well-known oncogene; its reactivation could give rise to transgene derived tumor formation in chimeric mice [31].
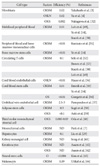
There have been several improvements of the gene transduction method for making safe iPSCs. Removal of the c-Myc oncogene from reprogramming cocktail is one of important approaches. Human and mouse iPSCs can be established from fibroblasts with only Oct4, Sox2, and Klf4, although the efficiency is significantly reduced [32]. Other many approaches have been designed to insert reprogramming factors into somatic cells (Table 1) [33-43]. One is the reduction of integration sites by putting the reprogramming factors into a single vector with an internal ribosome entry site or 2A self-cleavage peptide. This reprogramming cassette was used with a lentivirus system containing a loxP sequence in the long terminal repeat (LTR) and produced iPSCs with only single insertions [39]. The expression of Cre recombinase successfully cuts out the cassette. Although it leaves an incomplete LTR in the iPSC genome, this method minimizes the genomic alteration. A transposon system encoding a reprogramming factors cassette has also been successfully induced iPSC generation [37, 38]. The transduction of a plasmid-based transposon vector can integrate into the host genome with the help of transposase, and induces iPSC colony formation. The excision of the transposon does not leave a footprint, so it maintains the original endogenous sequences. Several other methods also accomplish iPSC generation by the transient expression of reprogramming factors. These include viral vectors (adenovirus and Sendai virus) [34, 35], DNA vectors (plasmid and episomal plasmid vector) [33, 36], or direct protein delivery [40]. Their efficiencies of iPSC induction are lower than that with retrovirus vectors, possibly due to low transduction efficiency, and unstable expression. A recent study used synthetic mRNA to reprogram human fibroblasts and differentiate into myogenic cells [42].
The mixture of specific reprogramming factors has been evaluated. The standard mixture contains Oct4, Sox2, Klf4, and c-Myc; this mixture has successfully induced cellular reprogramming in mouse, human, rat, pig and dog. Human iPSC induction has been achieved with a slightly different set of reprogramming factors, including Oct4, Sox2, Nanog, and Lin28 [33]. Inclusion of Oct4 and Sox2 in both sets indicates their importance for reprogramming. The reprogramming efficiency is enhanced by the addition of extra factors, such as ESRRB, UTF1, Sall4, Tbx3, mitochondrial RNAs (miRNAs, such as miR-291-3p, miR-294, and miR-295), and small hairpin RNAs (shRNAs) for p53 or p21. Lin28 and shRNA reprogramming factors for p53 mainly regulate the reprogramming efficiency through the control of cell proliferation [44]. Recently Anokye-Danso et al. [43] reported iPSCs can be generated solely through the expression of miR302/367. They show that miRNA-mediated reprogramming proceeds faster than with Yamanaka's four factor (Oct4, Sox2, Klf4, c-Myc) reprogramming.
Advances of hiPSCs Generation from Different Somatic Cell Types
One of the most important issues that hiPSCs can be applicable for clinical purposes is the generation of safe and functional cell types for cell based therapy. Mouse embryonic fibroblasts and tail-tip fibroblasts in mouse and dermal fibroblasts have been the cell types which are the most widely used to reprogram, because of their availability and easy accessibility. A comprehensive study using various mouse iPSCs have demonstrated that the origin of the iPSCs is very important on the tumor-forming propensities in a cell transplantation therapy model [45]. Mouse tail-tip fibroblast iPSCs (mesoderm origin) revealed as the highest tumorigenic propensity, whereas gastric epithelial and hepatocyte derived iPSCs (both are endoderm) have shown lower tumorigenic propensities [45]. Recent studies have suggested that mouse iPSCs of different origins possess distinct capacities to differentiate into blood cells [46, 47]. Although it has been demonstrated that hiPSCs retain certain gene expressions of the parent cells [48], it remains largely unclear whether the cell origin could affect the safety and function of hiPSCs. It is therefore extremely important to establish hiPSCs lines from multiple developmental origins and thoroughly examine the sources that impact on both the safety and their differentiation potentials. The ideal source of the cell to be isolated from the patients and used for reprogramming must have easy accessibility with minimal risk procedures, availability in large quantities, relatively high reprogramming efficiency, and fast iPSC derivation speed. Recent reports revealed that most of hiPSCs have been derived from mesoderm (fibroblasts and blood cells) or ectoderm (keratinocytes, melanocytes and neural stem cells) (Table 2) [3, 4, 7, 14, 29, 32, 49-64]. The technology to develop hiPSC lines provides a foundation to elucidate the mechanisms of cellular reprogramming and to study the safety and efficacy of differentially originated hiPSCs for cell therapy. The expression of the exogenous transcription factors may trigger a cascade of epigenetic events-chromatin modifications (e.g., DNA methylation, histone [de]acetylation), leading to iPSCs. It can be speculated that the modified chromatin state allows for easier access of the reprogramming factors to downstream genes needed for reprogramming [65].
Directed Differentiation of hiPSCs
One of the main strengths of the iPSC approach - the ability to generate large numbers of disease-specific iPSCs derived from the relevant cell types - can only be realized if a robust differentiation protocol for the desired cell type is available. For some cell types, differentiation is relatively well-defined. iPSCs can be readily differentiated into neurons, although the conditions to derive many specific neuronal subtypes remain unknown [17]. Cardiomyocytes can be easily obtained and identified [66]. Efficient differentiation protocols for other cell types, for example hepatocytes, are still being developed [29]. The hepatic differentiation of hiPSCs holds great promise as an ultimate source of hepatocyte which can be utilized for drug screening, disease modeling and cell therapy. However, more research is required to improve their differentiation efficiency and function of differentiated cells. The function of hiPSCs derived hepatocytes can be analyzed in vitro, by various methods, including analyses for cytochrome P-450 activity and glycogen storage ability with the periodic acid-Schiff assay [45]. Although these in vitro methods are highly informative and convenient, the most definitive proof for the function of hiPSCs derived hepatic cells would be the demonstration of hepatic engraftment in vivo using animal models [67] and detection of secreted human hepatocyte proteins in animal serum/plasma. A recent study demonstrated the feasibility of hiPSCs derived hepatocyte as modeling several inherited liver diseases [25]. Although in vitro culture may recapitulate certain disease features and may be suitable for drug screening purposes, successful regenerative therapy will require hepatic cells to be engrafted to the liver functionally. Even though hiPSCs can be differentiated to many lineages, overall remaining concern is to have safe cells and enough number of the iPSCs for the research as well as clinical trials.
Disease Modeling with hiPSCs
The concept of utilizing hiPSCs to model a disease in vitro is based on the unique capacity of these cells to continuously self-renew and their potential to give rise to all cell types in human body. Thus, hiPSCs could provide a limitless reservoir of cell types that, in many cases, would not be otherwise possible to obtain, for example, the motor and dopaminergic neurons affected in amyotrophic lateral sclerosis (ALS) and Parkinson's disease (PD). The overwhelming advantage of iPSC technology is that it allows for the generation of pluripotent cells from any individual in the context of his/her own particular genetic identity, including individuals with sporadic forms of disease and those affected by complex multifactorial diseases of unknown genetic identity, such as autism spectrum disorders [17] and type 1 diabetes [68]. Recently, a number of studies have reported the successful generation of patient-specific iPSC lines from individuals with any one of a number of diseases. However, effective disease modeling has been demonstrated in a few studies. For example, Ebert et al. [11] reported the differentiation of iPSC-derived motor neurons from a patient diagnosed with a genetic form of spinal muscular atrophy (SMA), a neurodegenerative disease that leads to loss of lower motor neurons. Importantly, this study was the first to demonstrate a disease-related in vitro phenotype in iPSC-derived cells. Motor neurons derived from the patient-specific iPSCs were initially similar in morphology and number to those derived from wild-type iPSCs. However, their numbers and size selectively declined after 8 weeks in culture. Furthermore, these cells exhibited a deficiency in survival of motor neuron (SMN) protein aggregates, which is a characteristic phenotype associated with SMA. Another study effectively demonstrated the potential of iPSC technology to model disease pathogenesis and treatment by creating iPSC lines from patients with familial dysautonomia (FD), a neuropathy caused by a point mutation in the iκB kinase complex-associated protein (IDBKAP) gene [12]. This mutation leads to a tissue-specific splicing defect that was recapitulated in iPSC-derived tissues. The authors went on to show disease-specific defects in neurogenesis and migration of neural crest precursors, tissues that were previously unobtainable. These disease-specific phenotypic changes were then assayed after treatment with candidate drugs, one of which had a beneficial effect. Recently, two groups reported generation of iPSCs from patients who have inherited liver diseases [69, 70]. Importantly, some of the key disease features of the inherited metabolic disorders were recapitulated in culture [70]. While the generation of disease-specific iPSCs is a critical first step, ultimately it will be derived to a representative set of hiPSCs from different patients.
Drug Screening/Drug Discovery Using hiPSCs
The costs of drug development are heavily influenced by compound attrition rate. For every drug that reaches the market, 5,000-10,000 compounds have been tested preclinically. More accurate predictive toxicity models would help reduce these costs. The hiPSCs also offers exciting opportunities for reliable high throughput drug screening in terms of specific disease phenotypes. This powerful ability in toxicology studies has the potential to increase the efficiency of novel human drug development, while reducing drug attrition in the final stages of development and therefore costs. Additionally, the use of iPSCs would also enable the single nucleotide polymorphism-related research that influences the ability of an individual to effectively metabolize and clear drugs and toxins. Accurate prediction of human drug toxicity is a key part of drug discovery process. In particular, hepatotoxicity and cardiotoxicity are two principal causes of drug failure during preclinical testing, while the variability in individual responses to potential therapeutic agents is also a major problem in effective drug development [71]. However, the safety evaluation process is hindered by the availability and quality of primary human liver models with which to study drug toxicity. The major hurdles in developing the scalable and high-fidelity human hepatocytes from hepatic cell lines, and fetal and adult progenitors have been limited organ availability, homogenous cell purification, short term cell culture, and rapid loss of hepatocyte phenotype and function in culture. The advantage of iPSC technology is that it allows the generation of a library of cell lines that may represent the genetic and potentially epigenetic variation of a broad spectrum of the population. Because hiPSCs can grow indefinitely in culture, they could provide the unlimited source for any desired specialized cells. Ultimately, the goal of this approach is to use an in vitro model of disease to identify novel drugs to treat the disease; for example, neurons of ALS and SMA patients or abnormal loss of insulin-producing β cells in diabetes patients. In fact, several laboratories have already derived iPSCs from patients of Huntington's disease, PD, ALS, juvenile diabetes, SMA, Fanconi's anemia and others [7, 8, 11, 19, 72, 73]. Moreover, promising reports showed that iPSCs derived from patients suffering from the devastating disorders SMA, FD and LEOPARD syndrome recapitulated the cell abnormalities an in vitro seen in patients [11, 12, 15]. Remarkably, with drugs for these diseases, the "symptoms" were partially alleviated in vitro. This principle can now be applied to many other diseases and cell types for which we currently do not have treatments, and may result in the development of drugs from which not just one individual, as in cell therapy, but many patients may benefit.
Therapeutic Potential of hiPSCs
An ultimate goal of iPSC research is using the iPSC (generation and differentiation) technology for cell therapy to intractable diseases. Because iPSCs can overcome the ethical issues related to ESC derivation and potential issues of allogenic rejection. This may represent a more ideal source to produce patient-specific and disease-specific adult cells for future clinical application and drug development. Organ transplantation among non-related individuals is very limited due to low availability of matched tissues and the requirement for life-long immunosuppressive drug treatment that can have serious side effects. hiPSCs can potentially circumvent these problems, as they could be coaxed into the desired cell types that would already be genetically matched with the patient. Another big advantage of iPSCs over current transplantation approaches is the possibility of repairing disease-causing mutations by gene targeting and other correction technologies. A proof of principle that iPSCs can be used to treat disease by correction of the underlying genetic defect was demonstrated in a mouse model of sickle cell anemia using gene editing method [16]. The wild-type β-globin gene was used to replace the defective gene by homologous recombination using zinc finger technology. Remarkably, transplantation with genetically corrected iPSC-derived hematopoietic progenitors was successful in ameliorating the symptoms of anemia and for restoring physiological function in the diseased animal. In principle, this approach could be applied to most of diseases in humans for which the underlying mutation is known, and that can be treated by cell transplantation. A similar approach was performed with human patients with Fanconi's anemia [8]. In this case, the mutant gene was replaced using lentiviral vectors prior to reprogramming of the patient's fibroblasts and keratinocytes, as the genetic instability of the mutant fibroblasts made them nonpermissive for iPSC generation. Importantly, these iPSCs could be differentiated into hematopoietic progenitors as efficiently as ESCs and wild-type iPSCs, stably maintaining the disease-free phenotype in vitro.
Gene Correction Methods for Personalized Medicine
The familial human disease is often linked to defined mutations in individual genes. Precise correction of these genetic lesions in patient-derived stem cells and iPSCs prior to in vitro differentiation and re-engraftment back into the donor is a critical barrier to the broad application of personalized autologous cell-based therapy. Classical gene targeting in mammalian cells combines positive and negative selection to isolate the rare targeted events [74]. Recent evolution of recombinant adeno-associated virus-mediated approaches has, however, boosted the efficiency of classical gene targeting in hiPSCs [75-77].
Zinc finger nuclease (ZFN) technology has emerged as a highly efficient new tool for precise eukaryotic gene editing directly at the endogenous genomic locus (reviewed by Urnov et al. [78]). ZFNs are comprised of a pair of engineered zinc finger DNA binding domains that are each linked to a modified catalytic nuclease domain of the restriction enzyme FokI [79-81]. The DNA binding specificity can be engineered to direct the ZFN pair to the desired genomic locus to induce a double strand DNA break (DSB) with high fidelity. The highly-conserved natural cellular DNA repair pathways of non-homologous end joining (NHEJ) or homology directed repair function to resolve the ZFN-induced DSB. NHEJ acts to efficiently rejoin the cleaved DNA ends. However, occasionally unfaithful repair can lead to variable small deletions or insertions at the site of DSB.
The recent application of ZFNs to genome editing in hiPSCs has signaled an important advance towards the goal of patient-derived cell-based therapy [82]. Initial studies used integration defective lentivirus (IDLV) to deliver both ZFNs and the donor repair template into hESCs to achieve targeted transgene insertion into the endogenous CCR5 locus in the absence of antibiotic selection [83]. Targeting of the same locus in human MSCs using adenoviral-mediated ZFN delivery coupled with IDLV-mediated donor delivery was also successful, again without selection [84]. While viral strategies can overcome the challenges of low delivery efficiency to recalcitrant cell types, optimized electroporation mediated nucleic acid delivery coupled with positive selection strategies have led to the successful inactivation of the endogenous genes, such as PIG-A, by insertion of a drug resistance expression cassette into the gene's coding sequence in both hESCs and hiPSCs [85].
Individual patient-derived iPSCs are providing new opportunities to modeling human disease in vitro [86]. Careful reprogramming of both wild type and diseased cells to become models for the tissues that are affected by the disease offers investigators a novel system for comparing the biology and, importantly, the drug sensitivity of affected and unaffected cells. However, notwithstanding the technical challenges of accurate and reproducible reprogramming, the molecular basis of a disease may differ between patients, even for apparently monogenic disorders. If two iPSC lines from the same disease behave differently it is difficult to determine whether the cause is other disease-relevant genetic differences, or a variable consequence of reprogramming. Generation of isogenic pairs of wild type and mutant iPSCs differing only in the disease-linked gene offers a solution resolving and eliminating variable genetic background as a confounding factor (Fig. 1). Yet to achieve this goal requires a strategy for gene correction in patient-derived iPSCs (or mutation of wild type cells) that leaves only the disease-linked mutation and no other genetic modification - including selectable markers used for isolating the genomic modification events or even the genetic "scar" left by recombinase-mediated removal of the marker. To this end, Soldner et al. [87] have used ZFN-based genomic editing to generate isogenic sets of human disease and control pluripotent stem cells that differ solely in the α-synuclein gene. Recent reports have shown that ZFNs can be used to efficiently insert both inducible and constitutively active constructs specifically into a safe harbor locus (AAVS1) in pluripotent stem cells to render their expression controllable by a small molecule drug [40, 88, 89]. Importantly, site-specific insertion into a safe harbor avoids the genomic and phenotypic uncertainty that surrounds random integration (e.g., epigenetic silencing of the transgene or disrupted regulation of other genes near the site of insertion), thereby providing the investigator with greater control over the biology of the cell.
Conclusion and Future Perspectives
Since the first description of iPSC generation, there has been remarkable progress toward clinical implementation of reprogramming technologies. However, iPSC-based therapies are still in their infancy, and many hurdles remain to be overcome before their clinical applications become a reality. The suitability of individual iPSC derivation methods for generating cell populations for cell replacement therapy, disease modeling, and drug discovery remains to be widely demonstrated, and studies assessing the equivalence of different types of iPSCs are eagerly anticipated. Moreover, extensive characterization of the functionality of iPSC-derived somatic cells and their functional equivalence with in vivo counterparts need to be widely demonstrated. The application of the benefits that iPSCs offer is also limited by the ability to derive disease-relevant somatic cells, and major challenges remain in defining pathways that efficiently lead to pure and functional populations of many disease-relevant cells. Given the rapid pace of developments within the iPSC field, it is likely that the future of personalized stem cell therapy will lie in our ability to take a patient's own cells, correct the disease allele, and then return those cells to the patient in a genetically and physiologically correct format (Fig. 2).


 XML Download
XML Download