

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor:
Steatocystoma multiplex (SM; OMIM184500) is a rare disorder of the pilosebaceous unit which is characterized by multiple sebum-containing dermal cysts. This disorder inherits an autosomal dominant mode, however, most sporadic cases have also been described. It may be associated with pachyonychia congenita, hypertrophic lichen planus, acrokeratosis verruciformis and so on1. Mutations in the keratin 17 gene (KRT17) underlie SM as well as pachyonychia congenita type 2. Thus, it is likely that these two conditions are phenotypic variants of the same disorder for some patients2,3.
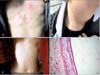
We reported a rare mutation of KRT17 in two patients from a Chinese SM family. The proband, a 22-year-old male, presented with many small cysts for 7 years. The cysts originally occurred in his chest, and then gradually involved other parts of his body and grew larger. On examination, numerous cysts with 0.1 to 0.5 cm in diameter were diffusely distributed around his entire body (Fig. 1A, B). All his fingernails and toenails were normal. His father also shared similar clinical features (Fig. 1C). The histopathology of biopsy taken from his upper chest showed flattened sebaceous lobules close to the cystic wall which consists of stratified squamous epithelia without a granular layer (Fig. 1D). Based on the clinical and histopathological features, the diagnosis of SM was established.
We collected blood samples and extracted genome DNA, and then carried out mutation analysis of KRT17 by direct sequencing when using the previous primers4 in all available family members and 676 unrelated healthy controls remained after informed consents.
The samples used for polymerase chain reaction were taken from the proband, his father and other family members as well as the 676 unrelated healthy controls. The two patients were confirmed by experienced dermatologists in the First Affiliated Hospital of Anhui Medical University. They had no abnormalities of nails. This study was approved by the Anhui Medical Institutional Review Board (2011937) and was conducted according to the Declaration of Helsinki Principles. The sequencing analysis showed a missense mutation of c.71C>T in the V1 domain of KRT17 gene in the proband and his father. This mutation resulted in change of the polar serine (Ser) residue at codon 24 into nonpolar leucine (Leu). It was not detected in unaffected members and in 676 unrelated healthy controls (Fig. 2).
We described two patients with typical clinical phenotype of SM and found a missense mutation c.71C>T of the KRT17 in one Chinese family. Interestingly, this mutation was located in the V1 domain, and not within the highly conserved regions as previous literatures described (http://www.interfil.org). The mutations outside the hotspot may lead to diseases by affecting the interactions between keratin and other protein molecules5.
In summary, we reported a novel missense mutation (c.71C>T) of KRT17 gene in one Chinese SM family. Further studies are required to elucidate the role of this mutation in the pathogenesis in SM.

 XML Download
XML Download