

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dermatophytes refer to a group of fungi that cause dermatophytosis by affecting keratinized tissues, such as the skin, hair, and nails of human and animal hosts, and include genera Trichophyton, Microsporum, and Epidermophyton1. Clinical manifestations of dermatophytosis vary depending on the site of infestation and the type of strain; therefore, accurate identification of the strain is crucial in order to facilitate rapid treatment and to prevent spread of the disease2. Especially now, with active participation in sporting events overseas and introduction of new exotic pets, new strains that have never been reported before are now being uncovered, and the demand for accurate identification of dermatophytes is higher than ever3-5. Previously, classification and identification of dermatophytes were conducted according to the clinical manifestation, gross examination of colonies from culture, microscopic examination of both macro- and micro-conidia, biochemical characteristics, and cross-culture6. However, this system of classification and identification is time-consuming, and may pose difficulties for non-experts in differentiation of the morphology of cultured colonies. Furthermore, even the same strains may show morphologically diverse colonies, making the identification of the causative organism more difficult. Therefore, exploration of the route of infection, differentiation between relapse and reinfection, variation of dermatophytes, and geographical distribution of strains is difficult to assess.
A number of studies have recently been reported, which reflects attempts to overcome these shortcomings and limitations of the conventional system of classification and identification, and to develop a faster and more precise system, with the help of various molecular biology techniques. Polymerase chain reaction (PCR) is quite handy and can be performed rapidly in general laboratories; consequently, many attempts are now being made to develop a more diverse array of both primers and reaction conditions for identification of dermatophytes. However, between individual species of dermatophytes, the differences in ITS1-2, 18S ribosomal RNA, and 28S ribosomal RNA are not significant, making the development of species-specific primers difficult. Nested PCR is valuable in that it is a rapid and reliable method for classification and identification of dermatophytes; however, its use for differentiation of species is quite often limited. The use of the PCR-DNA sequencing method may yield highly reliable results; however, its clinical application is often hampered by the length of time required and the cost7.
In this study, using 3 sets of specifically designed primers with multiplex PCR of our own design we isolated and identified the eleven standard species of dermatophytes. We also investigated whether multiplex PCR is indeed effective in identification of dermatophytes isolated from the scales of patients diagnosed with dermatophyte-related diseases.
MATERIALS AND METHODS
Materials
1) The standard strains
The study was carried out on the 11 standard strains of dermatophytes in the laboratory of the Department of Dermatology, Konkuk University School of Medicine (Table 1). The 11 strains included Epidermophyton floccosum var. floccosum (CBS 358.93), Trichophyton mentagrophytes var. mentagrophyte (CBS 126.34), Trichophyton tonsurans (CBS 483.76), Trichophyton verrucosum (CBS 134.66), obtained from the CBS Fungal Biodiversity Centre, Netherlands, and Trichophyton mentagrophytes var. interdigitale (IFM 53931), Microsporum audouinii (IFM5294 [CBS 408.51]), Microsporum canis (IFM 45829), Microsporum fulvum (IFM 5318), Microsporum gypseum (IFM 5292), Trichophyton violaceum (IFM 41075 [CBS 319.31]), obtained from the Chiba University research center, Japan, and, finally, Trichophyton rubrum (KCCM 60450 [ATCC 28188]), which were obtained from the Bioproduct Center, Biomedical Engineering Research Institute of Korea.
Methods
1) Culture of standard strains and extraction of DNA
The Sabouraud's dextrose agar (SDA) (i.e. dextrose 40 g, bacteriological peptone 10 g, bactoagar 8 g. Difco Laboratories, Detroit, MI, USA), sterilized at 121℃ for 15 minutes, was used for the medium. The 11 standard strains of dermatophytes were cultured onto the SDA at 32℃ for up to 2 weeks. Colonies cultured from SDA were sampled and transferred to a 1.5 ml e-tube and centrifuged at 12,000 rpm for ten minutes. The supernatant was removed, and 200µl-lysis buffer (100 mM Tris-HCl (pH 9.5)), 1 M KCl, 10 mM EDTA) was added; the colonies were then crushed with a plastic pestle. Proteinase K (Bioline, UK) 50 ng was added and the mixture was treated for 16 hours at 55℃, followed by 30 minutes at 100℃. The mixture was shaken thoroughly prior to addition of 400µl P/C/I (phenol: chloroform:isoamyl alcohol=25:24:1, v/v) and centrifuged at 13,000 rpm for 15 minutes at 4℃. The supernatant was removed, and an equal amount of isopropanol was added; DNA was concentrated at -80℃ for 1 hour. Purified DNA products were washed twice in 500 ml of 70% ethanol and centrifuged at 13,000 rpm for 5 minutes. The supernatant was removed and the DNA was dried at 37℃ for 20 minutes, dissolved in distilled water, and preserved at -20℃. The concentration of the extracted DNA was measured with the NanoDrop spectrophotometer ND-1000 (NanoDrop Technologies, Wilmington, DE, USA), and 50 ng was taken for use in PCR.
2) Direct extraction of the DNA from scales
The test tube containing scales, collected in the outpatient division, was centrifuged at 3,000 rpm for 10 minute, and as many scales as possible were transferred to a 1.5 ml e-tube. Scales that were difficult to handle were taken with a mineral oil-soaked cotton swab and only the cotton portion was cut out and transferred to a 1.5 ml e-tube. 100µl-lysis buffer (100 mM Tris-HCl (pH 9.5), 1 M KCl, 10 mM EDTA) was added and treated at 100℃ for 30 minutes. Proteinase K 50 ng was added and treated for 16 hours at 55℃, followed by 30 minutes at 100℃. After thorough mixing, 100µl P/C/I (phenol:chloroform:isoamyl alcohol =25:24:1, v/v) was added and centrifuged at 13,000 rpm for 15 minutes at 4℃. The supernatant was removed, and an equal amount of isopropanol was added; DNA was concentrated at -80℃ for 1 hour. Purified DNA products were washed twice in 500 ml of 70% ethanol, then centrifuged at 13,000 rpm for 5 minutes. The supernatant was removed and the DNA was dried at 37℃ for 20 minutes, dissolved in distilled water, and preserved at -20℃. The concentration of the extracted DNA was measured with the NanoDrop spectrophotometer ND-1000, and 50 ng was taken for use in PCR.
3) The primer design
Based on information gathered from the Blast database of NCBI, primers were designed using the Clustal W2 program, so that the 11 strains of dermatophytes could be distinguished from other pathogenic fungi, and that only dermatophytes would be amplified for analysis of both inter-specific similarities and differences, among the 11 strains (Table 2). The primer was produced at ITS1 (internal transcribed spacer 1) and ITS2 (internal transcribed spacer 2). Other sets of primers were developed at the sequence of 18S ribosomal RNA and 28S ribosomal RNA. The ITS1 and ITS2 primers were produced from the combination of forward 5'-ATCATTAACGCGCAGGC-3', reverse 5'-TGGCCACTGCTTTTCGG-3', the 18S ribosomal RNA primer from that of forward 5'-AAGTTGGTCAAACTCGGT-3', reverse 5'-TGATCCTTCCGCAGGTT-3', and the 28S ribosomal RNA primer from that of forward 5'-ACAGGGATTGCCCCAGTA-3', reverse 5'-CTTGTTCGCTATCGGTCTC-3'. The primer was solicited to and produced by Cosmo genetech (Cosmo genetech, Seoul, Korea).
4) Multiplex PCR
The reaction mixture of PCR amplification was adjusted to 50µl, which included 0.1 mM dNTPs, 10X PCR buffer, 0.5 mM primer, 0.6 U Taq polymerase (Enzynomics, Daejeon, Korea), and 50 ng of genomic DNA solution. The Veriti® 96-Well Fast Thermal Cycler (Applied Bio systems, Carlsbad, CA, USA) was used for PCR.
For the ITS 1 and 2, the reaction conditions were, as follows: 7 minutes of hot step at 94℃, one minute of denaturation at 95℃, one minute of annealing at 60℃, and one minute of extension at 72℃. The entire process was repeated for 35 cycles, with the final extension at 72℃ for 7 minutes. The PCR conditions for the 28S ribosomal RNA were, as follows: 7 minutes of hot step at 94℃, 1 minute of denaturation at 94℃, 30 seconds of annealing at 50℃, and 1 minute of extension at 72℃. The entire process was repeated for 35 cycles, with the final extension at 72℃ for 7 minutes. Finally, the PCR conditions of 18S ribosomal RNA were, as follows: 7 minutes of hot step at 94℃, 30 seconds of denaturation at 94℃, 1 minute of annealing at 57.5℃, and 1 minute of extension at 72℃. The entire process was repeated for 35 cycles, with the final extension at 72℃ for 7 minutes. The amplified DNA was observed on 3% (w/v) agarose gel in TAE buffer.
RESULTS
Identification of the standard strains using multiplex PCR
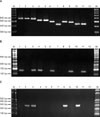
Results of PCR analysis from a dermatophyte specific primer (ITS1 and 2), the 18S ribosomal RNA primer set, and the 28S ribosomal RNA primer set of 11 strains of dermatophytes are shown in Fig. 1.
Bands obtained from the 11 strains, i.e., E. floccosum:M. audouinii:M. canis, T. rubrum:T. violaceum, T. mentagrophyte: T. interdigitale and M. gypseum:M. fulvum were located on 4 similar sites on PCR using a dermatophyte-specific (ITS1-2) primer (T. tonsurans and T. verrucosum were not). In order to further differentiate the strains which exhibited similar band patterns, PCR was performed using the 18S and 28S ribosomal RNA primers. Thus, a 50~80 bp-long band was visible on E. floccosum, T. mentagrophyte, T. violaceum, T. verrucosum, M. fulvum, and M. audouinii with the 18S ribosomal RNA primer PCR. In addition, a 300 bp-long band was identified with the 28S ribosomal RNA primer PCR on T. interdigitale, M. audouinii, M. canis and M. gypseum.
T. mentagrophytes showed differences between the two variant strains. Thus, T. mentagrophytes var. interdigitale exhibited a band in proximity to 300 bp on the 28S ribosomal RNA primer PCR, seen as 502 base pairs on the PCR analysis of ITS1-2. However, a band near 50 bp was identified in T. mentagrophytes var. mentagrophyte only on the 18S ribosomal RNA primer PCR, thus enabling differentiation of the two species. Also, a 50 bp-long band was visible only in T. violaceum on 18S ribosomal RNA primer PCR of T. rubrum and T. violaceum, each measuring 509 base pairs on ITS1-2. In the group of M. gypseum:M. fulvum, both exhibiting an approximately 470 base pairs band on ITS1-2, a 300 bp-long band was observed in M. gypseum on the 28S ribosomal RNA primer PCR, and a 100 bp-long band in M. fulvum on the 18S ribosomal RNA primer PCR.
Finally, a 300 bp-long band was observed in M. canis on the 28S ribosomal RNA primer PCR in E. floccosum:M. audouinii:M. canis, each with similar band patterns in proximity to a 560 bp band on ITS1-2, and a 100 bp-long and a 300 bp-long band in M. audouinii on the 18S ribosomal RNA primer PCR and 28S ribosomal RNA primer PCR, respectively. In E. floccosum, a 100 bp-long band was observed only on the 18S ribosomal RNA primer PCR.
Identification of clinical strains using multiplex PCR
Using multiplex PCR, the causative organisms were identified by extraction of the DNA directly from the scales of 73 outpatients, omitting the process of culture. Patients were stratified into groups according to the diagnosis, and the causative organisms were compared. T. rubrum was identified on multiplex PCR performed on scales from 24 patients with a clinical diagnosis of onychomycosis (Fig. 2). The causative agents of 19 cases of clinically diagnosed tinea pedis were T. mentagrophyte in one case, T. rubrum in 17 cases, with the other being T. tonsurans (Fig. 3). In 3 patients diagnosed with tinea capitis, T. rubrum was identified in 2 cases and T. tonsurans in the other (Fig. 4). In 16 tinea corporis patients, T. rubrum was found in 15 cases and M. gypseum was found in 1 case (Fig. 5). T. rubrum was identified in all 11 patients diagnosed with tinea cruris (Fig. 6).
Analysis of scales from 73 patients with fungal infection, irrespective of diagnosis, showed that T. rubrum was the most frequently identified species, with 69 cases with the exception of 1 case of T. mentagrophyte, 2 cases of T. tonsurans, and 1 case of M. gypseum (Table 3).
DISCUSSION
Traditionally, the process for identification of dermatophytes was carried out by examination of the shape of the colony and morphological features of hyphae and spores on microscopy. However, even within the same species, the morphology of dermatophytes may vary according to the conditions of culture, particularly in cases where subculture was performed for a long period of time. In addition, their typical features may be lost due to degeneration of villi. When morphological study is not feasible, additional tests, such as urease activity, vitamin dependency, hair perforation, and cross-culture may be used; however, they require highly skilled personnel, are time-consuming, and identification may still not be possible6,8. In order to solve these problems using a morphological method, various molecular biological techniques have been introduced from the early 1990's, and identification using restriction fragment length polymorphisms (RFLP), nested PCR, random amplification of polymorphic DNA (RAPD) analysis, and base-sequencing has been reported9-12.
The PCR method is highly sensitive and specific; therefore, detection of fungal cells is possible with as little as only 1 pg of DNA. This may also be applied to yeasts and other bacteria13. Multiplex PCR uses more than two primers for simultaneous amplification, and is widely used in the field of clinical microbiology, because it allows detection of a variety of microbes from a single specimen14. However, using the PCR method, both targeting of DNA to be amplified and selection of the primer are crucial. The primer should be designed with consideration for repeated base sequences that are common to all dermatophytes, random base sequences that possess a high probability of binding, and several specific base sequences within a single chromosome. The ITS1-2 primer which is commonly used in phylogenetic classification, is known to be the marker site for identification of dermatophytes species15. Although the phenotype and ecological characteristics of dermatophytes vary widely, depending on the type of strain, they are very similar genetically. In fact, upon sequencing of the 18S ribosomal RNA, 5.8S ribosomal RNA, and 28S ribosomal RNA regions, the difference in the number of base pairs between different strains is only 1 to 2, and possibly 5 or 6 at most. Therefore, perfect isolation and identification of the dermatophyte strain in a single session of PCR is very difficult. Noting both the similarities and differences in base sequence, the authors developed a total of 3 sets of primers, that include the ITS1-2, 18S ribosomal RNA, and 28S ribosomal RNA regions. Using these, we performed multiplex PCR on the 11 standard strains of dermatophytes and on the clinically obtained scales from patients. Analysis of the PCR results on the ITS1-2 regions of the 11 standard strains showed that all included a common base sequence, of approximately 400~600 bp in length. Also, differentiation between dermatophytes strains was partially successful with PCR analysis of the ITS1-2 region alone. These included only T. tonsurans (422 bp) and T. verrucosum (493 bp), with bands located at the lowermost area (Fig. 1). However, ITS1-2 alone was not sufficient for classification of dermatophytes. Thus, the 18S ribosomal RNA and 28S ribosomal RNA regions were included, thereby enabling us to categorize all of the strains to the genus level.
When dermatophytosis is suspected clinically, a KOH smear is done for simple confirmation of the presence or absence of fungus, or to carry out culture under appropriate temperature and humidity conditions for examination of the shape, color, or contour of the colonies, and to perform PCR. However, the culture process requires at least two weeks, and the fungi with fastidious culture conditions may not grow at all, or may be contaminated by other microbes. This problem may be prevented by direct extraction of DNA from the scales of patients, as in this study. Indeed, on multiplex PCR analysis of 73 patients with a clinical diagnosis of dermatophytosis, identification of the causative agent was completed in our experiments with the 3 sets of primers. The fact that the causative agent was T. rubrum in 95% of patients (69/73) is also in consensus with previous reports16. As evidenced, pinpoint identification is crucial from the therapeutic aspect, because Epidermophyton spp and Trichophyton spp are quite sensitive to terbinafine, whereas Microsporum spp are less sensitive17,18. Because treatment usually starts before identification of the organism, the role of PCR in clinical practice was not significant; however, the use of multiplex PCR may result in acceleration of early and precise identification, may facilitate treatment, and may predict both the clinical course and prognosis.
This is the first study to establish multiplex PCR using species-specific primers as a rapid and accurate method for identification of strains that cause dermatophytosis, from not only cultured colonies, but directly from the scales of patients, and we expect that it will be quite useful in the clinical practice.








 XML Download
XML Download