

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Depression is a common comorbidity in patients with epilepsy [1], but its treatment in these patients is challenging because many antidepressants induce pro-convulsive effects. Three major types of antidepressants—tricyclic antidepressant (TCA), selective serotonin reuptake inhibitor (SSRI), and serotonin-norepinephrine reuptake inhibitor—are known to increase seizure frequency although the pro-convulsive tendency varies among drugs [234]. TCAs induce seizures more frequently than the other antidepressants [234] and also increase arrhythmogenic risks [5678]. SSRIs induce pro-convulsive effects especially at high doses, but recent studies suggest that some SSRIs might counteract seizures at therapeutic doses in humans [9101112]. Given the broad use of SSRIs for treatment of depression, it is important to precisely determine their pro-convulsive risks and related mechanisms.
Paroxetine (Fig. 1), the most potent SSRI [1314], has lower toxicity than TCAs [1516] and lacks cardiovascular side effects [1516]. Pro-convulsive effects of paroxetine have been reported for humans and rats [171819], although a consensus on this effect is yet to be reached [22021]. Paroxetine modulates various types of ion channels related to neuronal excitability, via serotonin uptake-independent mechanisms. For example, paroxetine inhibits G protein-activated inwardly rectifying K+ channels (GIRK) [22] and TREK K+ channels [23], both of which contribute to hyperpolarizing the membrane potential of neurons [2425]. Thus, the paroxetine-induced inhibition of GIRK and TREK could render neurons more excitable. In addition to these channels, various types of ion channels are also involved in the modulation of spike frequency and membrane excitability. However, it is unclear whether and how paroxetine affects such diverse channels related to neuronal excitation.
Kv3.1 channels, one of the Shaw-type voltage-gated K+ channels, are abundantly expressed in GABAergic inhibitory neurons [262728] and related to the generation of fast, repetitive spikes [293031]. Kv3.1 channels are activated at depolarized membrane potentials and display rapid activation and deactivation kinetics [32]. Because Kv3.1 channels are implicated in the maintenance of fast spiking, their inhibition at depolarized potentials might delay membrane repolarization and thus dampen burst firing, as a simulation study displays [32]. The localization of Kv3.1 channels in interneurons suggests that a decrease in Kv3.1 current will enhance the net excitability of neural networks by weakening the activity of inhibitory neurons.
Fluoxetine, one of the widely used SSRIs, inhibits Kv3.1 channels [33] but it is unknown whether the inhibition of Kv3.1 channels is a common effect of SSRIs. Here, we hypothesize that paroxetine blocks Kv3.1 channels via direct interaction with channel pores. Our present data indicate that paroxetine acts as an open channel blocker of Kv3.1 channels, implying that paroxetine might contribute to the reduction in rapid firing of GABAergic interneurons in the brain.
METHODS
Cell culture and transfection
As previously described [3034], rat Kv3.1b cDNA [35] was subcloned into the expression vector pRc/CMV (Invitrogen Corporation, San Diego, CA, USA) and expressed in Chinese hamster ovary (CHO) cells. CHO cells were cultured in Iscove's modified Dulbecco's medium (IMDM; Invitrogen Corporation), supplemented with 10% fetal bovine serum, 0.1 mM hypoxanthine, and 0.01 mM thymidine, at 37℃ in a humidified atmosphere of 95% air and 5% CO2. The Kv3.1b expression vector was transfected into CHO cells using the Lipofectamine 2,000 reagents (Invitrogen Corporation). Transfected cells were selected in the presence of 500 µg/ml geneticin (A.G. Scientific, San Diego, CA, USA) and maintained in fresh IMDM containing geneticin. Following a brief trypsin/EDTA (Invitrogen Corporation) treatment, transfected CHO cells were seeded onto glass coverslips (12 mm diameter; Fisher Scientific, Pittsburgh, PA, USA) in a Petri dish before electrophysiological experiments. For electrophysiological recordings, coverslips with attached cells were transferred to a recording chamber (RC-13; Warner Instrument Corporation, Hamden, CT, USA), which was continuously perfused with the extracellular bath solution at a rate of 1 ml/min.
Electrophysiology
Potassium current through Kv3.1 channels was measured using the whole-cell patch-clamp technique [36]. All experiments were performed at 22–23℃. Ionic currents were amplified by a patchclamp amplifier (Axopatch 200B; Molecular Devices, Sunnyvale, CA, USA). Micropipettes were fabricated from glass capillary tubings (PG10165-4; World Precision Instruments, Sarasota, FL, USA) using a double-stage vertical puller (PC-10; Narishige, Tokyo, Japan) and had a tip resistance of 2-3 MΩ when filled with the intracellular pipette solution. For whole-cell recordings, the membrane capacitance and 80% of the series resistance were compensated by amplifier circuits. Leak subtraction was not used. The sampling rate was 5 kHz and the currents were low-pass filtered at 2 kHz through a four-pole Bessel filter. The generation of voltage-clamp pulses and data acquisition were controlled with pClamp 10.4 software (Molecular Devices) in a Windows-based computer interfaced to the amplifier by a Digidata 1440A acquisition board (Molecular Devices).
Solutions and drugs
The intracellular pipette solution for whole-cell recordings contained in mM: 140 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 EGTA (pH 7.3 with KOH). The bath solution for whole-cell recordings contained in mM: 140 NaCl, 5 KCl, 1.3 CaCl2, 1 MgCl2, 20 HEPES and 10 glucose (pH 7.3 with NaOH). Paroxetine (Fig. 1; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in ethanol at 30 mM and further diluted to working concentrations in the bath solution. The vehicle of paroxetine, <0.1% ethanol, had no effect on Kv3.1 currents (data not shown). To test the dose-dependency of paroxetine effect (Fig. 2), we applied a series of increasing concentrations of paroxetine (i.e., 3, 10, 30, and 100 µM) to a given cell.
Data analysis
Data were analyzed in the Origin 7.0 software (OriginLab Corp., Northampton, MA, USA). Interaction kinetics between the drug and channel was described based on a first-order blocking scheme as previously described [37]. From this concept, an IC50 value and a Hill coefficient (n) were obtained by fitting concentration dependence data (Fig. 2B) to the following equation:
in which I (%) is the percent current inhibition (I (%)=[1−Idrug/Icontrol]×100) at the test potential and [D] represents various drug concentrations.
The current activation curves (Fig. 3C) were fitted with the Boltzmann isotherm equation:
where V represents the test potential, V1/2 is the potential at which the conductance is half-maximal, and k is the slope factor; G is the conductance and Gmax is the maximal conductance. The conductance was calculated by G=I/(V−EK), where I is Kv3.1 current amplitude and EK is the calculated equilibrium potential of Kv3.1 (−84.6 mV).
To investigate the voltage dependence of Kv3.1 inhibition by the drug, the relative current amplitude was plotted as a function of the membrane potential (Fig. 3D). The resultant percent inhibition data between +30 and +70 mV were fitted with the Woodhull equation [38]:
where KD(0) represents the apparent affinity at 0 mV, z is the charge valence of the drug, δ is the fractional electrical distance, F is the Faraday's constant, R is the gas constant, and T is the absolute temperature. A value of 25.4 mV was used for RT/F at 22℃ in the present study. Equation 3 was modified for a linear transformation as follows.
The decay of Kv3.1 current (Fig. 4A) was fitted with a double exponential function:
in which τ1 and τ2 are the time constants; A1 and A2 are the amplitude of each exponential function; and B is the baseline constant.
The binding (k+1) and unbinding (k−1) rate constants were obtained from the following equations (Fig. 4B):
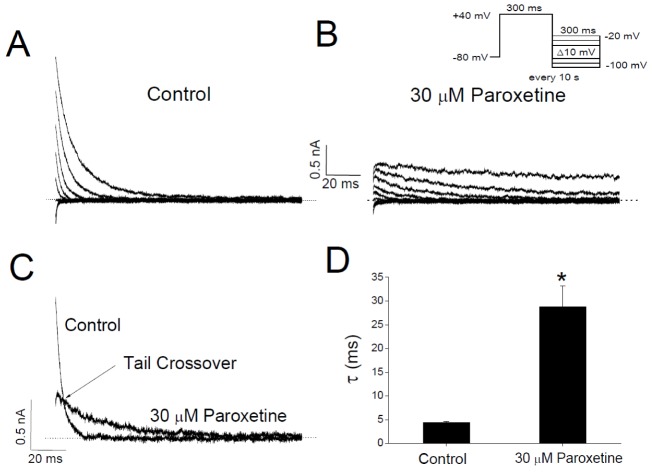
in which τD is the time constant of the drug-induced decay of Kv3.1 current. The time constant of Kv3.1 deactivation (Fig. 5) was obtained by fitting the tail current with a single exponential function (i.e., only one component of Equation 5).
Results were expressed as means±SEM. Student's t-test and analysis of variance (ANOVA) were used for statistical analysis. A two-tailed confidence level of p<0.05 was considered statistically significant.
RESULTS
Concentration-dependent and reversible inhibition of Kv3.1 channels by paroxetine
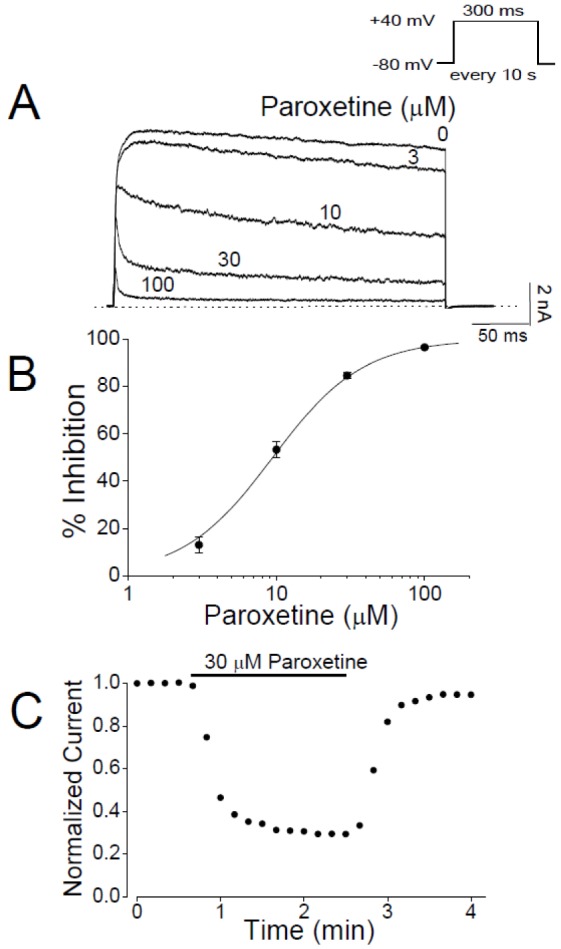
Potassium current through Kv3.1 channels, which were expressed in CHO cells, was recorded with the whole-cell patch clamp technique and its inhibition by paroxetine was tested. Kv3.1 channels were activated by 300-ms depolarizing pulses to +40 mV and paroxetine at 3 to 100 µM was applied to the extracellular solution (Fig. 2A). In the absence of paroxetine, Kv3.1 current rapidly activated and then slightly inactivated during the 40 mV pulse, as described previously [32]. In our control experiment, the whole-cell current recorded from non-transfected CHO cells contained no component of potassium current (data not shown) in accord with a previous report [39], suggesting that the Kv3.1 current recorded in our experiments was entirely generated by transfected channels. Paroxetine reduced the steady-state amplitude of Kv3.1 current and accelerated its decay in a concentration-dependent manner. Paroxetine suppressed the steady-state current of Kv3.1 by 13.0±3.4% at 3 µM and 96.6±0.2% at 100 µM (Figs. 2A and B). The concentration dependence of Kv3.1 inhibition by paroxetine was analyzed by a nonlinear least-squares fit of the current inhibition data with the Hill equation. The IC50 value and Hill coefficient were 9.43±0.53 M and 1.43±0.04 (n=4), respectively. As shown in Fig. 2C, the paroxetine effect on Kv3.1 current was induced rapidly and reached a steady state within 2 min after the onset of drug application. After paroxetine was washed out, Kv3.1 current recovered to 94.7±5.5% of the pre-drug baseline value within 2 min (n=4), indicating that effect of paroxetine was reversible.
Voltage-dependent inhibition of Kv3.1 by paroxetine
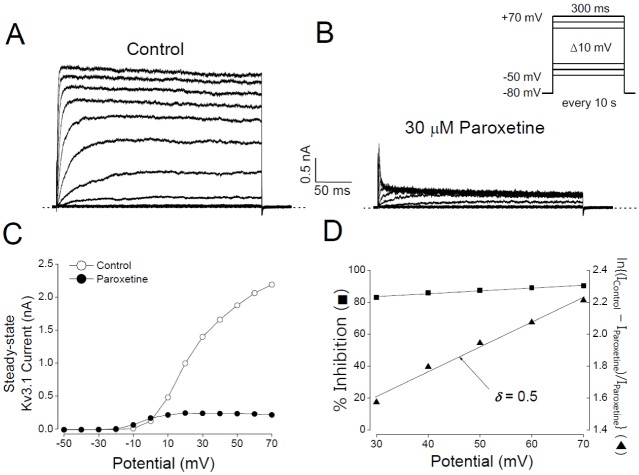
Next, we tested whether the decrease in Kv3.1 current by paroxetine was dependent on membrane potential. In the absence of paroxetine, the activation of Kv3.1 channels started at −20 to −10 mV (Figs. 3A and C). In additions, the steady-state I-V relationship showed a sigmoidal shape at potentials between −20 and +30 mV (Figs. 3A and C). Paroxetine (30 µM) inhibited Kv3.1 current in the entire voltage range over which Kv3.1 was activated, i.e., positive to −20 mV (Figs. 3B and C). When the degree of inhibition was plotted against activation voltage (Equation 3), a weak but significant voltage-dependent inhibition was observed over the voltage range where the channels are fully activated, i.e., +30 to +70 mV (Fig. 3D). In this voltage range, the magnitude of inhibition slightly increased with voltage: 82.8±4.8% inhibition at +30 mV and 90.1±5.9% at +70 mV (n=4, p<0.05). To estimate the site of action, we transformed the voltage dependence data of the paroxetine effect with the Woodhull equation and then fitted them with a linear line (Equation 4; Fig. 3D). The fitting indicated that the fractional electrical distance (δ) was 0.5±0.02 (n=4), implying that paroxetine might interact with Kv3.1 channels at the 50% point of the electric field of the membrane or channel pore. Furthermore, the positive sign of δ suggests that paroxetine is likely to act from the intracellular side of Kv3.1 channels.
Paroxetine action mechanisms determined by Kv3.1 current kinetics
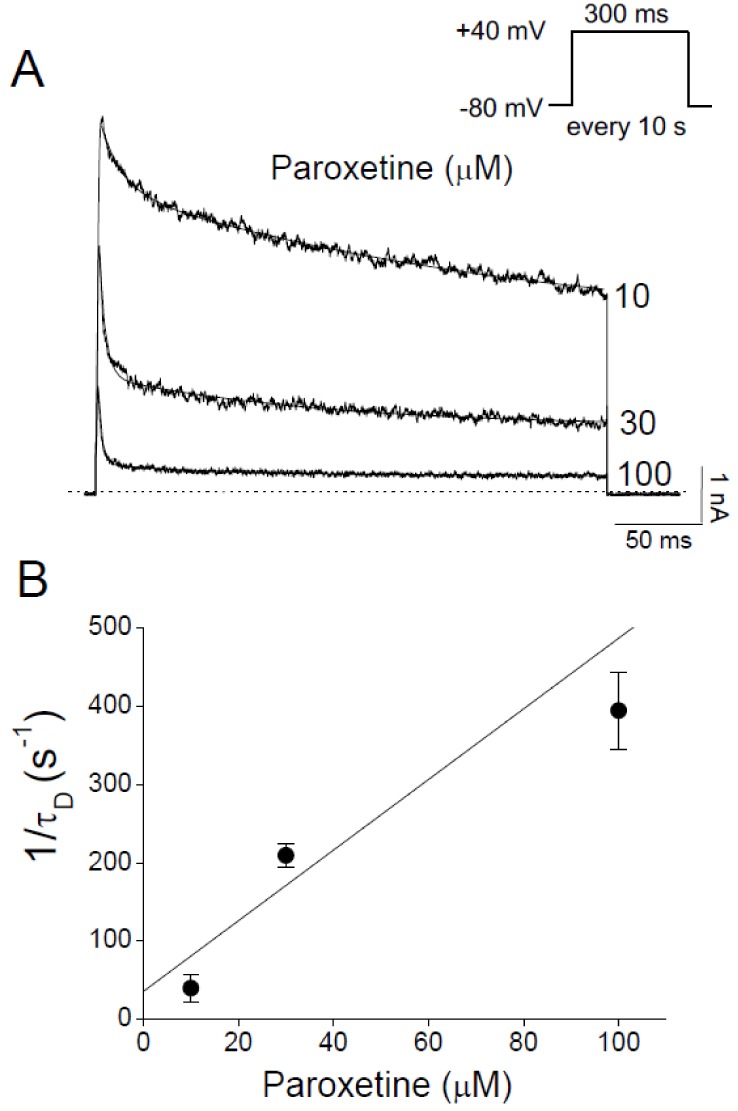
We investigated the kinetics of the inhibitory effect of paroxetine on Kv3.1 channels. Paroxetine accelerated the decay of Kv3.1 current in a concentration-dependent manner, resulting in biphasic decay of the current in contrast to the slow decay in the absence of paroxetine (Fig. 4A). This change in the decay pattern suggests that the fast decay of Kv3.1 current is likely to be induced by paroxetine. The time constant of each decay phase could be determined by fitting the Kv3.1 current with a double exponential function (Fig. 4A). The fast time constant for the decay of Kv3.1 current (τD) was taken as an estimate of the time course of the drug-channel interaction kinetics, whereas the slow time constant was considered to represent slow and partial inactivation, which is an intrinsic property of the Kv3.1 current [40]. The time constants were estimated in the presence of paroxetine at 10, 30, and 100 µM (i.e., when Kv3.1 current was decreased by >50%). To obtain the binding and unbinding rate constants of paroxetine, we plotted the reciprocal of τD at +40 mV against the concentration of paroxetine (Fig. 4B). This plot and Equation 6 yielded a binding rate constant (k+1) of 4.5±2.2 µM−1s−1 and an unbinding rate constant (k−1) of 35.8±7.1 s−1 (n=4). The KD value for open channel block calculated from k−1/k+1 (i.e., Equation 7) was estimated to be 7.9 µM, which is close to the IC50 value (9.43 µM) obtained empirically (Fig. 2A). The similarity between the KD and IC50 suggests that the drug and channel might interact with each other at a 1:1 ratio because our analysis of the kinetics was based on the assumption of 1:1 stoichiometry (see METHODS).
To further investigate the mechanism of paroxetine-induced inhibition of Kv3.1, we analyzed the effect of paroxetine on the time course of Kv3.1 deactivation. The deactivation kinetics was estimated from the Kv3.1 tail currents, which were activated by returning the membrane potential to various levels (−100 to −20 mV) after Kv3.1 activation at +40 mV (Figs. 5A and B). At the repolarizing pulse of −40 mV, for example, the tail current under the control condition declined quickly with a time constant of 4.37±0.29 ms (n=4) when fitted with a single exponential function (Figs. 5C and D). However, in the presence of 30 µM paroxetine, the initial peak amplitude of the tail current was markedly reduced and the subsequent decline of the current was slower (τ=28.77±4.42 ms; n=4; p<0.05), resulting in the tail crossover phenomenon (Figs. 5C and D). Paroxetine (30 µM) did not change the reversal potential of Kv3.1 tail current, which was about −84 mV either with or without paroxetine (Figs. 5A and B), indicating that paroxetine has no effect on the ion selectivity of Kv3.1 channels. The slow deactivation with single exponential kinetics and the crossover of tail currents support the idea that paroxetine might block the open state of Kv3.1 channels (see DISCUSSION).
Use-dependent inhibition of Kv3.1 channels by paroxetine
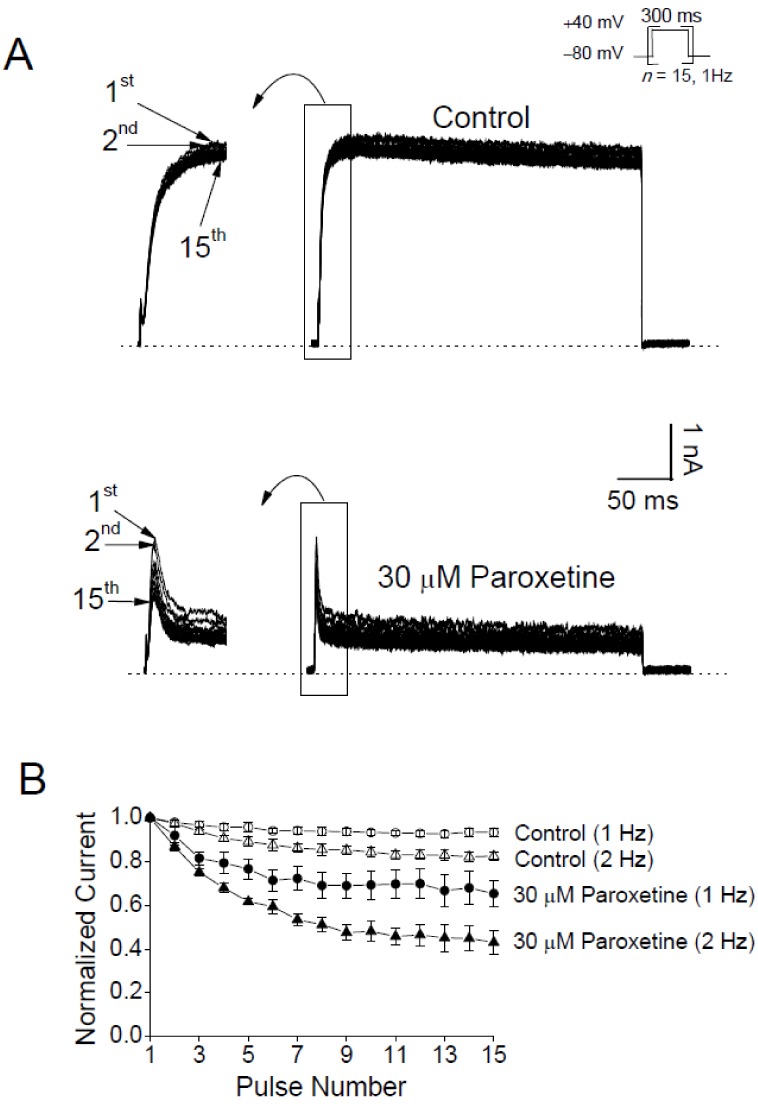
If paroxetine blocks the open pores of Kv3.1 channels as our analyses suggest (Figs. 3, 4, 5), paroxetine is predicted to inhibit the channel in a use-dependent manner. To test this prediction, we repetitively evoked Kv3.1 current with +40 mV pulses 15 times at 1 or 2 Hz (Figs. 6A and B) in the absence and presence of paroxetine (30 µM). Without paroxetine, the peak amplitude of the 15th Kv3.1 current at the end of the 1 Hz train slightly decreased by 6.7±1.8% (n=4) compared to the amplitude of the first current in the train. In the presence of paroxetine, Kv3.1 currents displayed higher levels of use-dependent reduction compared to the non-paroxetine control group: the peak amplitudes of Kv3.1 currents were progressively decreased by a significantly larger magnitude, 34.7±5.9% (n=4; p<0.05), at the end of the 1 Hz train. The result of greater use-dependence with paroxetine suggests that paroxetine might act as an open channel blocker. Such an effect of paroxetine was also observed when Kv3.1 was activated at 2 Hz. At the end of the 2 Hz train (i.e., at the 15th pulse), the peak amplitude of Kv3.1 current was decreased by 17.6±1.7% (n=4) and 66.9±5.6% (n=4) in the absence and presence of paroxetine, respectively (p<0.05; Fig. 6B). It is interesting to note that the use-dependent inhibition with 2 Hz stimulation was more pronounced than that with 1 Hz activation. This result is also consistent with the idea that paroxetine might block the open channel of Kv3.1.
DISCUSSION
The present study demonstrates that paroxetine, an SSRI antidepressant, inhibits the Kv3.1 potassium channels by acting as an open channel blocker. We examined the effect of paroxetine using the whole-cell patch clamp technique on Kv3.1 channels cloned from rat neurons and expressed in CHO cells. Although some SSRIs, including paroxetine, have been reported to induce or enhance epileptic activity in the brain [234171819], the involved mechanisms are unclear. Because Kv3.1 channels play an important role in the generation of high-frequency, repetitive action potentials in GABAergic interneurons, the paroxetine-induced reduction in Kv3.1 current might contribute to dampening interneuronal firing, and hence, increasing the overall excitability of neural networks.
The following lines of evidence from our data strongly support the idea that paroxetine preferentially interacts with the open pores of Kv3.1 channels resulting in their blockade. (1) Paroxetine accelerated the decay of Kv3.1 current during a depolarizing pulse, implying that paroxetine is likely to block the channels in their open state. Facilitated decay of current is commonly observed with other open channel blockers [33414243444546]. (2) Paroxetine inhibited Kv3.1 current in the entire voltage range over which Kv3.1 channels were activated. This result suggests that Kv3.1 channels need to be open for the inhibitory effect of paroxetine to occur. (3) Paroxetine slowed the time course of channel deactivation, which was assayed as tail current. The consequent phenomenon of tail crossover suggests an interaction between paroxetine and Kv3.1 channels in the open state [41424347]. (4) The inhibitory effect of paroxetine on Kv3.1 channels was use-dependent, i.e., enhanced at higher rates of channel activation. This result is consistent with the effects of open channel blockers [4143].
It is noteworthy that the paroxetine-mediated inhibition of Kv3.1 channels increased with membrane potential even when the channels were fully activated, i.e., over the voltage range positive to +30 mV (Fig. 3D). This voltage dependence of the inhibition provides an important insight into the site of paroxetine action within the transmembrane electric field. At physiological pH (either extracellular or intracellular pH 7.2–7.4), a majority of paroxetine is positively charged because the drug is a weak base with a pKa value of 10.32 [48]. If paroxetine acts from the extracellular side of the channel, the inhibition of Kv3.1 channels should be less pronounced as the membrane becomes more depolarized because positive membrane potential repels the positively charged drug. However, our result demonstrates that the voltage dependence of the paroxetine effect was in the opposite direction (Fig. 3D), implying that paroxetine is likely to move into the transmembrane electric field from the intracellular side. The δ value of 0.5 estimated from the voltage dependence indicates that the positively charged paroxetine senses 50% of the applied transmembrane electrical field as referenced from the intracellular side. This δ value is larger than those previously obtained with open channel blockers of Kv1.3 (δ=0.29), Kv1.1 (δ=0.25) [4749], Kv1.5 (δ=0.16–0.19) [42445051] and Kv3.1 channels (δ=0.31–0.38) [3346]. Therefore, the location of interaction between paroxetine and Kv3.1 appears to be deeper with respect to the intracellular side than those between other open channel blockers and Kv channels mentioned earlier.
Kv3.1 channels are characterized by a high activation threshold and very rapid activation and deactivation kinetics [3252]. Based on the high activation threshold, i.e., about −10 mV (Fig. 3C), it has been suggested that Kv3.1 should be activated in the late phase of action potential and might play a key role in repolarizing the membrane potential following the peak of action potential. By facilitating membrane repolarization, Kv3.1 channels could contribute to shortening the intervals between action potentials, resulting in increases in spike frequencies [2753]. In addition, the fast deactivation of Kv3.1 could quickly decrease K+ conductance after action potentials and thus reduce the refractory period of spikes. All of these properties of Kv3.1 channels might be related to the fast and repetitive generation of action potentials. Indeed, a computer simulation study has shown that modifications of Kv3.1 activity would affect neuronal excitability by altering the duration and frequency of action potentials [32].
A decrease in Kv3.1 current might reduce neuronal firing, but its consequence in neural networks must be evaluated based on the cellular loci of Kv3.1 channels. For example, firing of pyramidal neurons and interneurons will increase and decrease, respectively, the network excitability. Kv3.1 channels are expressed in inhibitory, but not excitatory, neurons in the brain [262728] and involved in high-frequency firing of interneurons [293031]. Therefore, blockade of Kv3.1 channels could reduce interneuronal spiking, resulting in weakened GABAergic inhibition and hence an increase in network activity. Because our data indicate that the inhibition of Kv3.1 channels by paroxetine is use-dependent, the suppressive effect of paroxetine, if any, on fast spiking of interneurons could become more pronounced during repetitive firing. The close resemblance in biophysical properties between cloned Kv3.1 channels and the neuronal endogenous Kv3.1 channels [293031] suggests that the paroxetine effects observed in the present study could also be replicated in intact neurons, but this issue is yet to be tested.
It should be noted that the net effects of paroxetine on neural networks will be determined by combinations of various factors, such as the location of other paroxetine-sensitive ion channels and the potency or efficacy of paroxetine for other types of channels. The IC50 values of paroxetine are 14-203 µM for GIRK [22] and 5.5 µM for TREK K+ channels [23], implying that paroxetine might also inhibit these channels at concentrations effective on Kv3.1. The inhibition of GIRK and TREK will elevate the excitability of both glutamatergic and GABAergic neurons, but given the large population of excitatory neurons (~90% of neurons) [54], the net effect is likely to be an increase in network excitability. Paroxetine also inhibits Nav1.4 and Nav1.7 sodium channels, which are required for the generation of action potentials, with the IC50 values of 10–28 µM [555657]. Nav1.4 and Nav1.7 channels are primarily expressed in the muscle and nociceptors, respectively [58], and therefore, paroxetine-mediated inhibition of these channels might not reduce network excitability in the brain.
Because interneuronal activity suppresses paroxysmal network excitation, the blockade of Kv3.1 (e.g., by paroxetine or fluoxetine), and hence reduced firing of interneurons, could perhaps elevate epileptic activity. If an antidepressant also possesses proconvulsive effects, extra caution is required when it is used for patients with both depression and epilepsy. Future studies should be directed toward the detailed characterization of the extent and strength of pro-convulsive activity of SSRIs including paroxetine. Specifically, it needs to be tested whether (1) paroxetine indeed induces epileptic effects in live animals and humans; (2) the inhibition of Kv3.1 channels is a common effect of not only paroxetine or fluoxetine but also other SSRIs; and (3) other ion channels or signaling cascades are also involved in the pro-convulsive effects of SSRIs.
In conclusion, the present study describes, for the first time, the effects of paroxetine on the Kv3.1 channels cloned from rat neurons and expressed in CHO cells. Detailed analyses of the interaction kinetics between paroxetine and Kv3.1 suggest that paroxetine blocks Kv3.1 channels in an open state in concentration-, voltage-, and use-dependent manners, implying that paroxetine might suppress fast spiking of interneurons, and thus, increase the overall network excitability.
 XML Download
XML Download