

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Microglia are macrophage-like cells in the brain and they are strongly implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer's disease [1,2] and Parkinson's disease [3]. Microglia are activated by inflammatory and pathophysiological stimuli in neurodegenerative diseases [1], and activated microglia increase phagocytic functions by releasing cytokines, chemokine, reactive oxygen species, and other immune factors [4,5]. When microglia are activated by lipopolysaccharide (LPS), production of inducible nitric oxide synthase (iNOS) is upregulated, leading to enhanced generation of nitric oxide (NO), which may induce neuronal damage [6,7,8,9]. Thus, understanding the mechanisms regulating microglial activation is important to reduce neuronal damage that is aggravated by activated microglia in neurodegenerative diseases.
Vitamin D3 is a steroid hormone that is important in the regulation of plasma calcium concentration. The ultraviolet exposure of 7-dehydrocholesterol present in the skin results in the photochemical production of vitamin D3, which is biologically inert. The liver enzyme 25-hydroxylase converts vitamin D3 into 25-hydroxyvitamin D3 [25(OH)D3, cholecalciferol], the biologically active form of vitamin D3. In the kidney proximal tubule, 1-α-hydroxylase converts 25(OH)D3 into 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], the hormonally active vitamin D metabolite [1,10]. Immune-related cells are also reported to transform 25(OH)D3 to 1,25(OH)2D3 [11,12,13]. Both the vitamin D receptor (1,25-dihydroxyvitamin D3 receptor, VDR) [14,15,16] and 1-α-hydroxylase [15] are widely expressed in neuronal and glial cells in the brain. Although microglia express both VDR and 1-α-hydroxylase, resting microglia produce barely detectable amounts of 1,25(OH)2D3 [17] while activated microglia synthesize significant amounts of 1,25(OH)2D3 from 25(OH)D3 in the rat brain [17], which suggests a possible role of vitamin D3 in microglial activation.
Some reports have suggested an effect of vitamin D3 on neuroinflammation and microglial activation. 1,25(OH)2D3 inhibits the expression of iNOS in the rat brain during experimental allergic encephalomyelitis [18] and after intracranial injection of LPS [19]. It also reduces the production of proinflammatory cytokines and NO in stimulated EOC13 microglial cells [20]. Pretreatment with 1,25(OH)2D3 decreases 6-hydroxydopa-induced dopaminergic neuronal injury by preventing microglial activation [21]. A recent study examining the effect of dietary vitamin D on microglial activation indicated that high dietary doses of vitamin D3 could attenuate microglia activation in a toxic model of demyelination [22]. Although most studies about the effects of vitamin D3 were performed using 1,25(OH)2D3, the plasma concentration of 25(OH)D3 is an indicator of adequate intake of dietary vitamin D3 in many vitamin D-related clinical conditions [23,24]. Thus, to elucidate the possible role of vitamin D3 as a negative regulator of activated microglia in neuroinflammatory conditions, the effect of 25(OH)D3 on activated microglia should be investigated.
In this study, we have found that 25(OH)D3 inhibited the production of NO in LPS-activated microglia through the mediation of LPS-induced 1-α-hydroxylase, and that 25(OH)D3 exerted its inhibitory effect by preventing LPS-induced phosphorylation of p38 through the mediation of vitamin D-VDR signaling.
METHODS
Materials
LPS, p-nitrophenyl phosphate, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The antibody for β-actin was obtained from Santa Cruz Biotechnology (Paso Robles, CA, USA). Antibodies for p38, p-p38 and VDR were supplied by Cell Signaling (Danvers, MA, USA).
Culture of primary and BV2 microglial cells
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Kyung Hee University and all efforts were made to minimize the number of animals utilized as well as animal suffering. The forebrains of newborn Sprague-Dawley rats were cut into small pieces and cells were dissociated by mechanical disruption using nylon mesh. The cells were seeded into T-75 flasks. After culturing in vitro for 10~14 days, microglial cells were isolated from mixed glial cultures by mild trypsinization (0.05% trypsin). The purity of microglial cultures was greater than 95% as determined by isolectin B4 staining (data not shown). The immortalized murine BV2 microglial cells [25] were kindly provide by Dr. Sun-yeou Kim (Gachon University, Incheon, Korea). The BV2 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin-streptomycin.
Nitric oxide and cell viability assays
In order to determine the level of NO production, BV2 cells were plated into 96-well plates and treated with LPS (100 ng/ml) in the presence or absence of 25(OH)D3 (1~100 nM) for 24 h. The concentration of nitrite, a soluble oxidation product of NO, in the media was determined using the Griess reaction. The supernatant (50 µl) of each well was harvested and mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% N-1-napthylethylenediamine dihydrochloride in 5% phosphoric acid). After 10 min, the absorbance at 540 nm was measured using a microplate reader. Sodium nitrite was used as a standard to calculate NO2 concentration. Cell viability was measured using the MTT assay.
siRNA-mediated knockdown of gene expression
siRNA reagents against 1-α-hydroxylase and VDR were purchased from Santa Cruz Biotechnology (Paso Robles, CA, USA). BV2 cells (5×104) were plated in 24-well plates and transfected with 1-α-hydroxylase (20 µmol/L) or VDR (50 µmol/L) siRNA using DharmaFECT reagent (Dharmacon Inc, Chicago, IL, USA) according to the manufacturer's protocol. After 24 h, the culture medium was replaced with fresh supplemented medium, and the cells were cultivated for an additional 24 h.
RNA extraction and cDNA production
Total RNA was extracted using TRIzol (Gibco BRL, Gaithersburg, MD, USA) and subjected to RT-PCR using the following primers: 5'-CAGAGGCAGCCATGAGGAAC-3' (sense) and 5'-GGGTCCCTTGAAGTGGCATAG-3' (antisense) for 1-α-hydroxylase; 5'-ATGGGTCAGAAGGACTCCTACG-3' (sense) and 5'-AGTGGTACGACCAGAGGCATAC-3' (antisense) for β-actin. The cDNAs were amplified by 28 cycles at 55℃ for 45 s on a thermocycler (GeneAmp PCR System9700, Applied Biosystems, Carlsbad, CA, USA).
Western blotting
Cells were lysed in a lysis buffer [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1 mM phenylmethylsulfonyl fluoride]. Protein concentration was determined using a Bio-Rad protein assay kit. An equal amount of protein for each sample was separated by 8% SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membrane was blocked with 5% non-fat milk and incubated with the primary antibody (VDR, p38, p-p38, or β-actin). After removing the primary antibody, the membrane was washed and incubated with horseradish peroxidase-conjugated secondary antibody. The immune-reactive proteins were detected using an ECL reagent (Thermo Scientific, Chicago, IL, USA).
RESULTS
Inhibitory effect of 25(OH)D3 on the production of NO in LPS-activated microglia
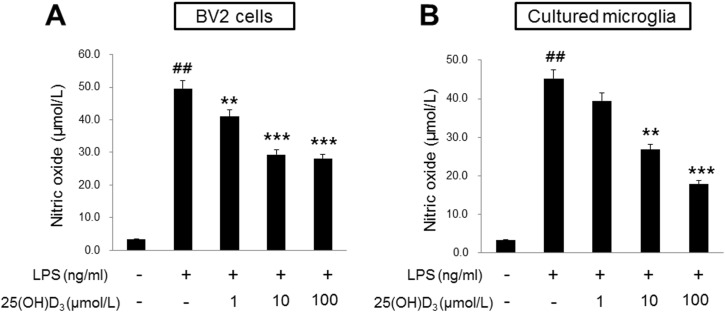
To examine the effect of 25(OH)D3 on the production of NO in LPS-activated microglia, we measured the amount of NO production in BV2 cells and primary cultured microglia that had been activated by LPS treatment (Fig. 1). After treatment with 100 ng/ml LPS for 24 h, the NO level in BV2 cells was 49.6 µmol/L (Fig. 1A). Production of NO in LPS-treated BV2 cells was significantly reduced in the presence of 10 and 100 µmol/L of 25(OH)D3 (29.3 and 28.0 µmol/L of NO, respectively; Fig. 1A) but was not affected in the presence of 1 µmol/L of 25(OH)D3. In primary cultured microglia, treatment with 100 ng/ml LPS for 24 h increased the production of NO (45.2 µmol/L, Fig. 1B). Treatment with 25(OH)D3 at a concentration of 10 and 100 µmol/L inhibited the production of NO in LPS-treated primary microglia (26.7 and 17.8 µmol/L, respectively; Fig. 1B). Cell viability was assessed using the MTT assay to rule out the possibility that the decreased NO production might be due to cell death. After BV2 cells were treated with 25(OH)D3 at concentrations of 1, 10, and 100 µmol/L for 24 h, the cell survival rate was more than 90%. This confirms that observed effects on NO production were not caused by cell death (data not shown). These results indicate that activated microglia might be suppressed by 25(OH)D3.
Effect of 1-α-hydroxylase siRNA on the inhibitory effect of 25(OH)D3
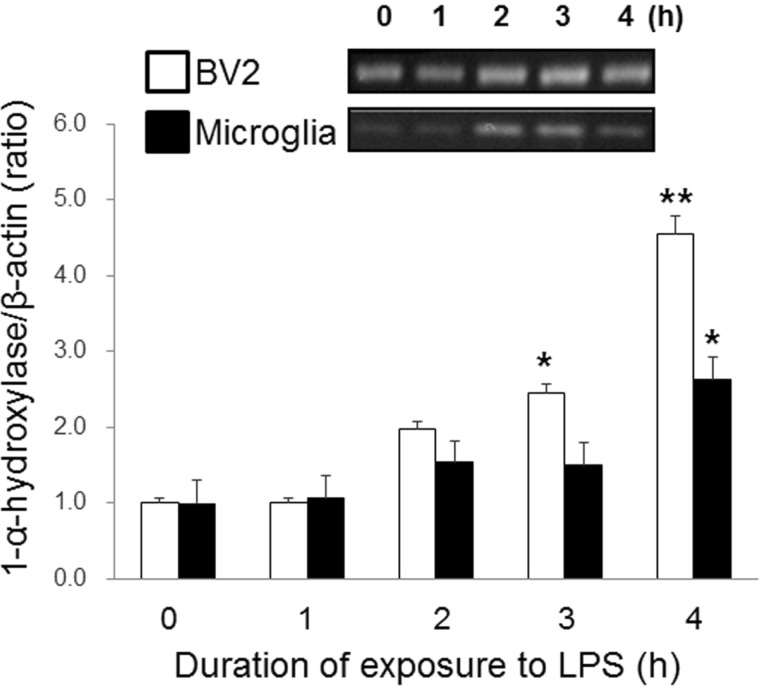
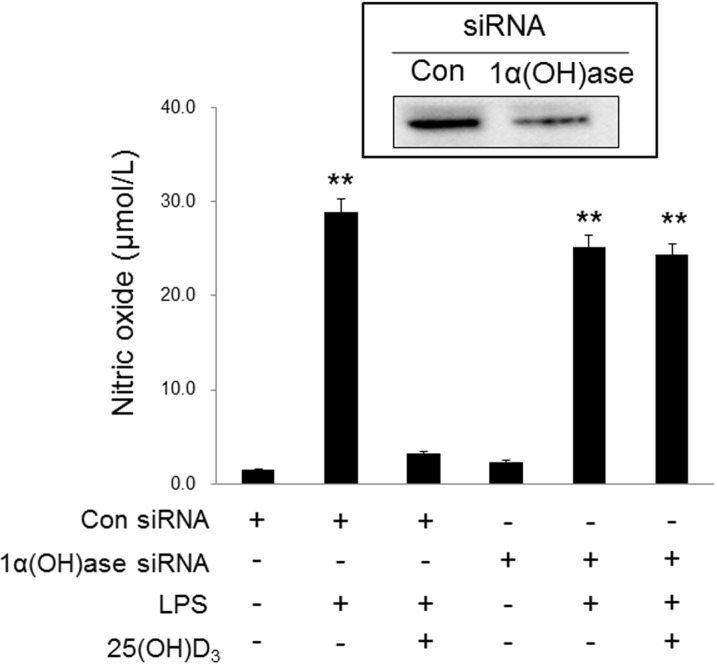
An enzyme that converts 25(OH)D3 to 1,25(OH)2D3, 1-α-hydroxylase, is widely distributed in microglia [15]. 1,25(OH)2D3 has been reported to inhibit activated microglia [18,19,20,21]. In this study, we postulated that the inhibitory effect of 25(OH)D3 on LPS-activated microglia (Fig. 1) could have resulted either from a direct effect of 25(OH)D3 itself or from the effects of 1,25(OH)2D3 that has been converted from 25(OH)D3 by 1-α-hydroxylase. To differentiate between these two possibilities, we examined the effect of 1-α-hydroxylase siRNA on the inhibitory action of 25(OH)D3 in LPS-activated microglia. Fig. 2 shows that 1-α-hydroxylase is expressed in BV2 cells and primary cultured microglia and that treatment with 100 ng/ml LPS for more than 3~4 h significantly increased the expression of 1-α-hydroxylase. After the expression of 1-α-hydroxylase was blocked with 1-α-hydroxylase siRNA, the effect of 25(OH)D3 on the LPS-activated microglia was examined (Fig. 3). In BV2 cells treated with a control siRNA, treatment with LPS (100 ng/ml) for 36 h increased the production of NO (28.8 µmol/L). Treatment with 100 µmol/L 25(OH)D3 decreased the production of NO (<10 µmol/L) in LPS-activated BV2 cells. However, while treatment with 1-α-hydroxylase siRNA did not affect the LPS-induced increase in NO levels (25.2 µmol/L), the inhibitory effect of 25(OH)D3 was completely suppressed (24.3 µmol/L of NO) in LPS-activated BV2 cells. These results indicate that LPS-induced up-regulation of 1-α-hydroxylase might be essential to the inhibitory effect of 25(OH)D3 on LPS-activated microglia.
Effect of vitamin D receptor siRNA on the inhibitory effect of 25(OH)D3
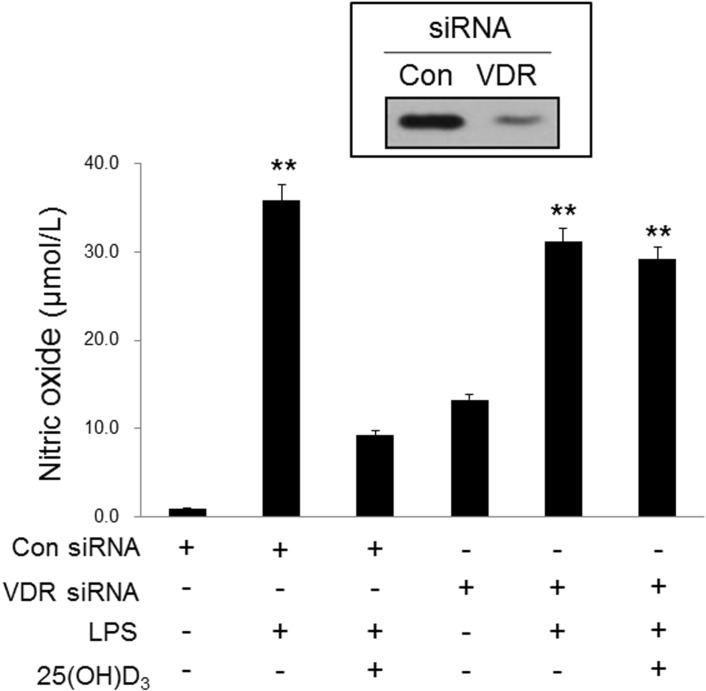
Glial cells, including microglia, have been reported to express VDR receptors in their nucleus [14,15]. In this study, 25(OH)D3 inhibited LPS-activated microglia in a manner that was dependent on the hydroxylation into 1,25(OH)2D3 by 1-α-hydroxylase. Thus, the 1,25(OH)2D3 receptor VDR might be important for the inhibitory effect of 25(OH)D3 on LPS-activated microglia. To elucidate the importance of VDR, we examined the effect of VDR knockdown on the inhibitory action of 25(OH)D3 in LPS-activated microglia. Treatment of BV2 cells with 100 ng/ml LPS for more than 1 h significantly increased the expression of VDR (data not shown). After the expression of VDR was blocked with VDR siRNA, the effect of 25(OH)D3 on the LPS-activated microglia was examined (Fig. 4). In BV2 cells treated with control siRNA, treatment with LPS (100 ng/ml) for 36 h increased the production of NO (35.8 µmol/L). Treatment with 100 µmol/L 25(OH)D3 decreased the production of NO (9.2 µmol/L) in LPS-activated BV2 cells. When the expression of VDR was blocked with VDR siRNA, the LPS-induced increase in the production of NO was not significantly altered (31.2 µmol/L). However, in LPS-activated BV2 cells treated with VDR siRNA, the inhibitory effect of 25(OH)D3 on the production of NO was completely suppressed (29.1 µmol/L). These results indicate that 25(OH)D3 exerts its inhibitory effect on LPS-activated microglia in a VDR-dependent manner.
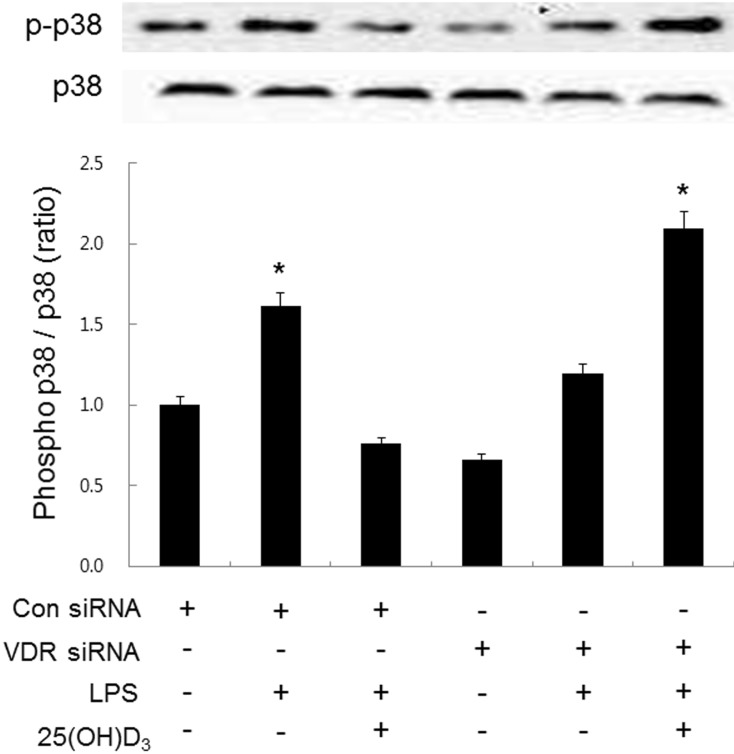
Effect of 25(OH)D3 and VDR siRNA on the p38 phosphorylation in LPS-activated microglia
It is well known that LPS-treated microglia induce inflammation through phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) [26,27,28,29,30,31]. Experiments performed in macrophages, adipocytes, prostate cells, and podocytes have shown that vitamin D exerts an anti-inflammatory effect by inhibiting p38 MAPK phosphorylation through VDR signaling [32,33,34,35]. The present study has shown that LPS-induced up-regulation of VDR is important to the inhibitory effect of 25(OH)D3 on LPS-activated microglia. We therefore hypothesized that p38 MAPK phosphorylation might be involved in the inhibitory effect of 25(OH)D3 on LPS-activated microglia. To elucidate this possibility, we examined the effect of 25(OH)D3 on LPS-induced p38 phosphorylation in BV2 cells treated with VDR siRNA (Fig. 5). When BV2 cells were treated with LPS (100 ng/ml, for 36 h), the expression ratio of phosphorylated p38/total p38 was increased 1.7-fold. Treatment with 25(OH)D3 (100 µmol/L) inhibited the increased phosphorylation of p38 MAPK in LPS-activated BV2 cells. When BV2 cells were transfected with VDR siRNA (50 µmol/L), the inhibitory effect of 25(OH)D3 on the LPS-induced phosphorylation of p38 MAPK completely disappeared. These results indicate that 25(OH)D3 reduces LPS-induced p38 phosphorylation through VDR signaling and inhibits the inflammatory response of LPS-activated microglia.
DISCUSSION
Microglia are activated by inflammatory stimuli in neurodegenerative diseases [1], and activated microglia increase their phagocytic function by releasing reactive oxygen species and other immune factors [4,5]. Activated microglia synthesize a significant amount of vitamin D3 in the rat brain [17] and vitamin D3 has an inhibitory effect on activated microglia [18,19,20,21]. Thus, it has been suggested that vitamin D3 plays a role as a negative regulator of activated microglia. In this study, the regulatory effect of 25(OH)D3 on LPS-stimulated microglia was investigated. We have found that the increased NO production in LPS-activated microglia is inhibited by 25(OH)D3 in a manner that is dependent on the LPS-induced 1-α-hydroxylase. We also showed that 25(OH)D3 exerts its inhibitory effect by inhibiting LPS-induced phosphorylation of p38 through VDR signaling.
Most studies showing the inhibitory effect of vitamin D3 on activated microglia were performed using 1,25(OH)2D3 [18,19,20,21]. However, 1,25(OH)2D3 is formed from the hydroxylation of 25(OH)D3 by an enzyme 1-α-hydroxylase [1,10,20,26]. Additionally, the plasma concentration of 25(OH)D3 is an indicator of adequate intake of dietary vitamin D3 in vitamin D-related clinical conditions [23,24]. Thus, the effect of 25(OH)D3 on activated microglia should be investigated in order to clearly understand the mechanism of action of dietary vitamin D3 on microglia-induced neuroinflammation in neurodegenerative diseases. In the present study, 25(OH)D3 inhibited the NO production of LPS-activated microglia (Fig. 1). Because the 1-α-hydroxylase enzyme is widely expressed in glial cells of the brain [15], the inhibitory effect of 25(OH)D3 could be a direct effect on activated microglia or an indirect effect through the conversion into 1,25(OH)2D3. In this study, LPS-stimulated microglia increased the expression of 1-α-hydroxylase (Fig. 2) and treatment with 1-α-hydroxylase siRNA completely blocked the inhibitory effect of 25(OH)D3 on the NO production of LPS-activated microglia (Fig. 3). These results suggest that 1-α-hydroxylase in activated microglia converts 25(OH)D3 into 1,25(OH)2D3, which then inhibits NO production. However, further study might be necessary to measure the production of 1,25(OH)2D3 in LPS-activated microglia to clearly elucidate whether 25(OH)D3 is converted into 1,25(OH)2D3.
After being produced from 25(OH)D3, 1,25(OH)2D3 binds VDR and initiates vitamin D-VDR signaling. Previous studies performed in multiple cell types have shown that vitamin D exerts an anti-inflammatory effect by inhibiting the phosphorylation of p38 MAPK through VDR signaling [32,33,34,35]. Activated microglia induce inflammation through the phosphorylation of p38 MAPK [26,27,28,29,30,31] and VDR is widely expressed in microglia [14,15]. Thus, it is possible that the inhibitory effect of 25(OH)D3 on activated microglia might occur by inhibiting LPS-stimulated p38 phosphorylation through VDR signaling. In this study, the inhibitory effect of 25(OH)D3 on activated microglia was completely blocked by VDR siRNA (Fig. 4). 25(OH)D3 completely blocked the LPS-induced phosphorylation of p38 MAPK and this blocking effect of 25(OH)D3 was completely abolished by VDR siRNA (Fig. 5). These results suggest that 25(OH)D3 reduces LPS-induced p38 phosphorylation through VDR signaling, thus inhibiting the inflammatory response of LPS-activated microglia. However, it is possible that other kinases might be involved in the effect of 25(OH)D3 on LPS-activated microglia. Thus, p38 might be not the only kinase but one of the kinases involved in the regulatory effect of 25(OH)D3.
It is controversial whether activation of microglia is detrimental or beneficial to neuronal cells. However, it is clear that sustained and strong inflammatory reactions mediated by microglia can trigger neuronal cell damage. Thus, it has been suggested that a negative regulatory mechanism might be present to restrict the process of microglial activation. According to the results of the present study, activated microglia might increase the expression of 1-α-hydroxylase and VDR. 25(OH)D3 is converted into 1,25(OH)2D3 by 1-α-hydroxylase, which then stimulates VDR signaling and inhibits the phosphorylation of p38 in activated microglia. This cascade might inhibit the inflammatory reaction of activated microglia. In conclusion, the present study suggests that vitamin D3 might have an important role in the negative regulation of microglial activation.
 XML Download
XML Download