

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The principal role of vascular smooth muscle cells (VSMCs) in blood vessels is to mediate the contractions in response to extracellular stimuli, such as angiotensin II (AngII) [1]. Contractions from AngII stimulation is mainly acquired by maintaining its contractile phenotype [2]. For example, the proliferation of fully differentiated VSMCs stops and the expression of contractile proteins, such as myosin heavy chain (MHC), myosin light chain kinase (MLCK), calponin, smooth muscle actin (SMA) and SM22α, remains at a high level to provide contractility [1]. Although skeletal muscle cells lack the ability to proliferate and migrate, VSMC has plasticity in their phenotypes [1]. The contractile phenotype of VSMC can undergo a phenotypic change, thereby retaining their ability to proliferate in response to environmental stimuli [3]. The significance of phenotypic switch is often attained in many vascular diseases, such as atherosclerosis, arteriosclerosis, restenosis, and vascular aging [3]. Hence, unveiling the mechanism of phenotypic change seems to be a key step in understanding many vascular diseases.
Krüppel-like factors (KLFs) belong to transcription factors containing zinc finger domains, and they regulate a variety of physiologies, such as differentiation, development, and proliferation [4]. Thus far, there have been 17 human KLF proteins. KLFs can be classified into three groups by multiple sequence alignments and phylogenic analyses: Those that are in Group1 (KLF3, KLF8, KLF12) suppresses transcription through the interaction with the carboxy-terminal binding protein (CtBP); those in Group2 (KLF1, KLF2, KLF4, KLF5, KLF6, KLF7) serve as transcriptional activators or repressors; and those in Group3 (KLF9, KLF10, KLF11, KLF13, KLF14, KLF16) have repressor activity through an interaction with transcriptional corepressor Sin3A [5].
Transcriptional activity of KLFs is modulated by a variety of mechanisms. For example, cAMP response element binding protein (CBP) and p300 acetylates KLF4 and KLF5, facilitating transcriptional activity [67]. Conversely, histone deacetylase 1 (HDAC1) interacts with KLF5, thereby dissociating KLF5 from p300, which blocks the KLF5-dependent target gene expression [8]. An interaction of KLFs with other proteins is also affected by phosphorylation. For instance, all-trans retinoic acid induces phosphorylation of HDAC2, disrupts the interaction between KLF4 and KLF5, allowing KLF5 to be acetylated and bind to SM22α promoter in VSMCs [9]. The transcriptional activity of KLFs can directly be regulated by phosphorylation. For instance, a stimulation of VSMCs with AngII directly induces the phosphorylation of KLF5, modulating the transcriptional activity of KLF5 [10]. The transcriptional activity of KLFs can also be regulated by modulating the expression levels through ubiquitination and sumoylation [11]. Serum stimulation of HCT116 cancer cells rapidly degrades the KLF4 level through ubiquitination and proteasome pathway [12]. Likewise, it has been reported that expression level of KLF5 is regulated by ubiquitin-dependent proteosomal degradation [13]. In addition to the ubiquitin-dependent regulation of KLF4 and KLF5, expressions of KLF4 and KLF5 are also regulated by sumoylation. KLF4 and KLF5 interact with the SUMO E3 ligase PIAS1 [14]. In case of KLF4, the degradation of KLF4 by sumoylation stimulates the promoter activity of SMA [15]. However, sumoylation of KLF5 stimulates nuclear translocation, which promotes the gene expression of cell cycle regulator [16].
A variety of environmental cues—inflammation, diabetic condition, hypoxic condition, and malnutrition—stimulates VSMCs, which affects vascular integrity [17]. Previously, many reports suggest that tumor necrosis factor α (TNFα) plays a crucial role in vascular remodeling. For example, the loss of TNFα reduces the thickness of vascular walls and the size of atherosclerotic lesions in TNFα/ApoE double knockout mice [1819]. However, the mechanistic pathway underlying TNFα-induced phenotypic conversion of VSMCs is still ambiguous.
In this present study, we explore the role of KLFs during the TNFα-induced phenotypic conversion of VSMCs and provide evidence that myocardin, KLF4, and Akt1 regulates transcriptional activity of KLF8 which in turn suppresses transcriptional activity of KLF5, thereby enhancing the expression of contractile marker protein of VSMCs.
METHODS
Antibodies against SMA, calponin, and SM22α were obtained from Sigma-Aldrich (St Louis, MO, USA). Anti-actin antibody was purchased from MP Biomedicals (Aurora, OH, USA). Anti-KLF5 antibody was purchased from EMD Millipore (Billerica, MA, USA). AngII and laminin, isolated from the human placenta, were obtained from Sigma-Aldrich (St Louis, MO, USA). Type I collagen was purchased from BD Biosciences (San Jose, CA, USA). Recombinant human TNFα was obtained from KOMA Biotech (Seoul, Korea). IRDye700- or IRDye800-conjugated rabbit or mouse secondary antibodies were obtained from Li-COR Bioscience (Lincoln, NE, USA). Plasmids that encode KLF4 and KLF5 as well as pGL3-KLF5 were kindly provided by Professor Mukesh K. Jain (Case Western Reserve University School of Medicine). FLAG-tagged KLF4, KLF5, KLF8, Akt1, and myocardin were PCR amplified and subcloned into pMIGR2 vector at EcoRI/BamHI. The promoter regions of SM22α and SMA subcloned in pGL3 vector were kind gifts from Dr. Gary K. Owens (University of Virginia). pGL3-KLF8(Δ0) and its deleted mutants (Δ1~Δ4) were kindly provided by Professor Jihe Zhao (Albany Medical College). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise indicated.
Cell culture and phenotypic conversion
VSMCs were isolated from 4-week-old Sprague-Dawley rats, as described previously [20]. In brief, rats were euthanized via intraperitoneal injection of sodium pentobarbital (60 mg/kg) and perfused with PBS for 5 min. The thoracic aorta was aseptically isolated and the surrounding fat and connective tissues were discarded. The vessels were longitudinally cut, and the lumen side was scraped with a razor blade to remove the intima. The vessels were then fragmented into 3~5 mm lengths and explanted with the lumen side down on collagen-coated culture dishes. After seven days of maintenance in Dulbecco's modified Eagle medium (DMEM) containing 10% bovine calf serum and 1% penicillin-streptomycin at 37℃ in a humidified 5% CO2 incubator, the tissue fragments were discarded, and sprouted VSMCs were collected (referred to as P0). Synthetic VSMCs were cultured on gelatin-coated plates at low density (<20%). To acquire the contractile VSMCs, synthetic VSMCs (P0) were cultured on laminin-coated plates at high density (~100%), and passages between P2 and P5 were defined as the contractile phenotype of VSMCs. The phenotype of VSMCs was verified by Western blotting with SMC marker proteins, such as SM22α, calponin, and SMA. To induce the phenotypic conversion of VSMCs, TNFα (50 ng/ml) was supplemented in a culture medium, which was changed on a daily basis, for a total duration of 4 days.
Promoter assay
For the measurement of promoter activity, the dual-luciferase reporter assay system was employed. VSMCs were plated in 12-well plates. The cells were co-transfected with the luciferase reporter constructs and renilla luciferase plasmid, using Lipofectamine 2000 (Invitrogen). Each well contained 0.88 µg of luciferase reporter plasmid, 0.8 µg of expression vector, and 80 ng of renilla luciferase plasmid. The medium was replaced with a fresh medium at 7 h post-transfection. The cells were lysed and assayed for luciferase activity at 24 h post-transfection. Twenty microliters of protein extracts were analyzed in a Glomax™ 20/20 luminometer (Promega, WI, USA).
Collagen gel contraction assay
Collagen gel contraction assay was performed as described previously [20]. VSMCs were starved for 4 h and resuspended in serum-free DMEM (1×106 cells/ml). Cell suspension was mixed with collagen gel solution (8 mg/ml of collagen type I in 2X PBS, pH 8.0) on ice to give 5×105 cells/ml and 4 mg/ml of collagen gel solutions. One hundred microliters of VSMC-collagen gel mixture was added to 12-well plates. The plates were incubated at 37℃ to allow for polymerization. After 30 minutes, the gels were floated in serum-free DMEM; and after 5 hours, AngII was added to initiate a contraction while images were captured using a digital charge-coupled device camera. Collagen gel contraction was measured as a decrease in the gel area using Scion Image software (compliments of Scion Corporation, Frederick, MD; http://www.scioncorp.com). Relative gel area was obtained by dividing the area at each time point by the initial area of the gel.
Constructs and primers
To silence the genes of interest, oligonucleotides tagged with 5′-end AgeI site and 3′-end EcoRI site were designed for Akt1 (5'-CCG GTA ACT TCT CAG TGG CAC AAT GCC TCG AGG CAT TGT GCC ACT GAG AAG TTT TTT TG-3'), and both sense and antisense oligonucleotides were synthesized. Both complementary oligonucleotides were mixed and heated at 98℃ for 5 min, and then cooled to room temperature. Annealed nucleotides were subcloned into the AgeI/EcoRI site of a pLKO.1 lentiviral vector. To measure the expression of KLF8, sense (5′-ACG CCC CAG GTG GAA CCA GT-3′) and anti-sense (5′-TGG CTG CAG GGC TCC CAT CT-3′) oligonucleotides were synthesized and RT-PCR was performed as previously described [20].
Lentiviral knockdown
For gene silencing, HEK293-FT packaging cells were grown to ~70% confluence in 6-well plates. The cells were triple transfected with 5 µg of pLKO.1 lentiviral construct, 1 µg of Δ8.9, and 1 µg of pVSV-G using the calcium phosphate method. The medium was replaced with a fresh medium at 8 h post-transfection. Lentiviral supernatants were harvested at 24 h post-transfection and passed through 0.45 µm filters. Cell-free viral culture supernatants were used to infect the contractile VSMCs in the presence of 8 µg/ml of polybrene. An additional round of infection was done at 48 h and 72 h post-transfection. The infected cells were isolated by 10 µg/ml puromycin for 2 days.
RESULTS
Down regulation of KLF8 during TNFα-induced phenotypic change of VSMCs
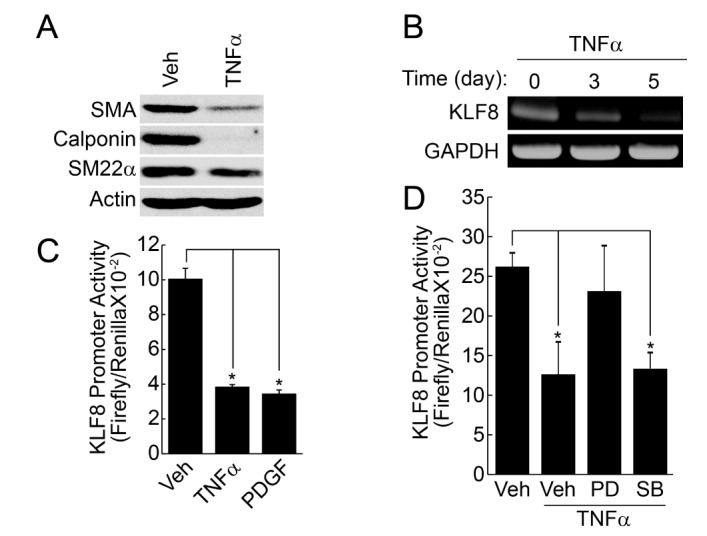
Stimulation of contractile type of VSMCs with TNFα (50 ng/ml) significantly reduced the expression of marker proteins, such as SMA, SM22α, calponin (Fig. 1A). In addition, the expression of KLF8 was also significantly reduced by TNFα in a time-dependent manner, as judged by RT-PCR. The promoter activity of KLF8 was also reduced by the pretreatment of TNFα and platelet-derived growth factor (PDGF) (Figs. 1B and 1C), which is known as an inducer of phenotypic change [21]. TNFα-induced down regulation of KLF8 promoter activity was significantly blocked by the inhibition of ERK signaling pathway, whereas the inhibition of p38 MAPK pathway had no effect (Fig. 1D).
Regulation of KLF8 promoter activity by KLF4 and Akt1
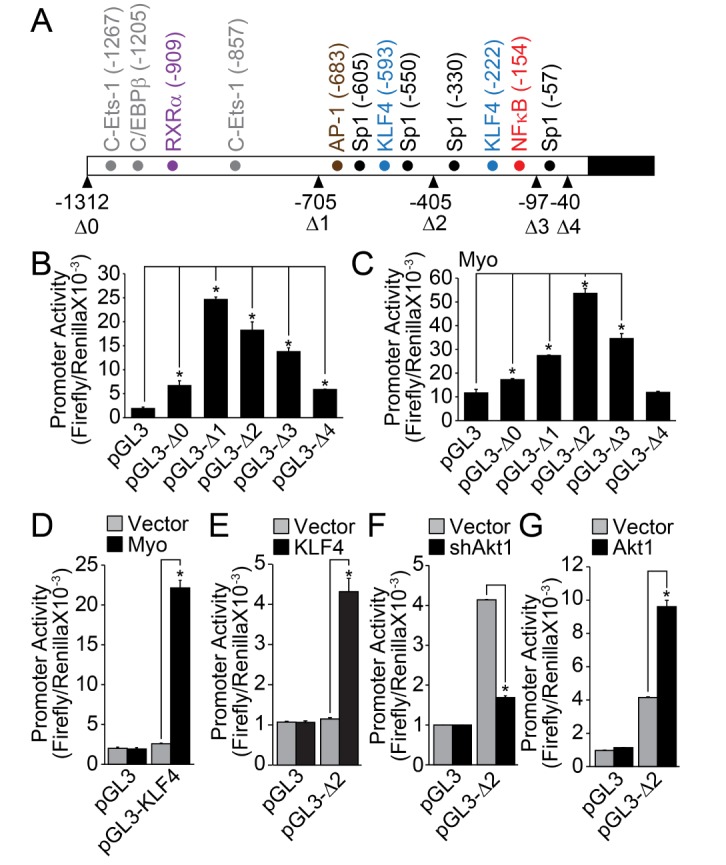
The analysis of KLF8 promoter revealed that many cis-acting elements, such as C-Ets-1, CAAT/enhance binding protein β (C/EBPβ), retinoic acid receptor α (RXRα), Ap1, Sp1, KLF4, and NFκB, were located in the promoter region (up to – 1312) (Fig. 2A). Deletion up to –705 markedly enhanced the promoter activity of KLF8, indicating the presence of negative regulatory region between –1312 and –705. Moreover, deletion of last Sp1 binding site (–57, Fig. 1A) completely abolished the promoter activity of KLF8, suggesting that Sp1 transcription factor regulates the basal promoter activity of KLF8. The co-transfection of myocardin markedly enhanced the promoter activity of KLF8, especially in the form of pGL3-Δ2 (contains KLF4 and NFκB binding sites). This indicates that myocardin regulates KLF8 expression through the regulation of KLF4 and/or NFκB (Fig. 2C). Myocardin significantly stimulated the promoter activity of KLF4 (Fig. 2D), and the co-transfection of KLF4 significantly enhanced the promoter activity of KLF8 (Fig. 2E). Finally, silencing or overexpression of Akt1, which has been known as the upstream regulator of NFκB, significantly affected the promoter activity of KLF8 (Figs. 2F and 2G).
Expression of KLF8 significantly upregulates VSMC marker gene expression
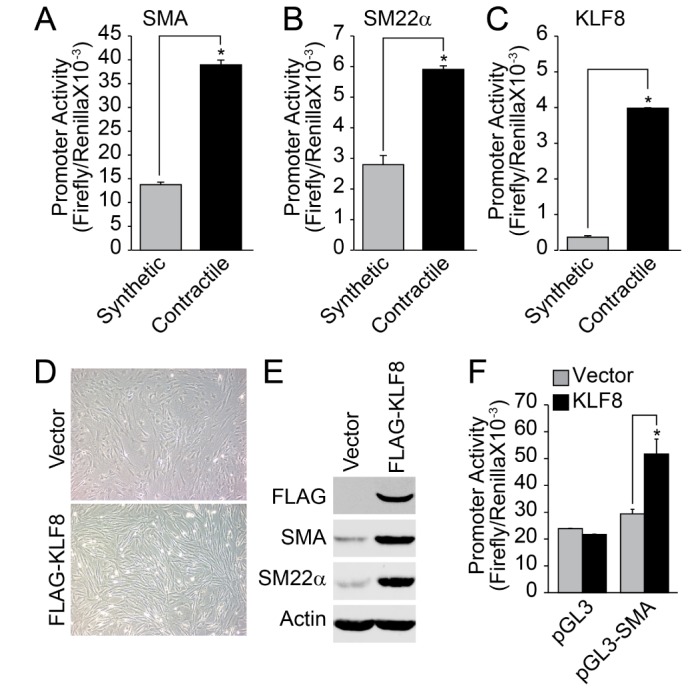
The promoter activity of VSMC marker genes, such as SMA and SM22α, was significantly enhanced in the contractile phenotype of VSMCs (Figs. 3A and 3B). Additionally, the promoter activity of KLF8 was also enhanced in the contractile phenotype of VSMC (Fig. 3C). Fibroblast-like morphology of synthetic VSMC was changed into an elongated spindle shape by the overexpression of KLF8 (Fig. 3D). In addition, the expression of contractile marker genes was significantly enhanced by the overexpression of KLF8 (Fig. 3E). Finally, the overexpression of KLF8 strongly enhanced the promoter activity of SMA (Fig. 3F).
KLF8 regulates promoter activity of KLF5
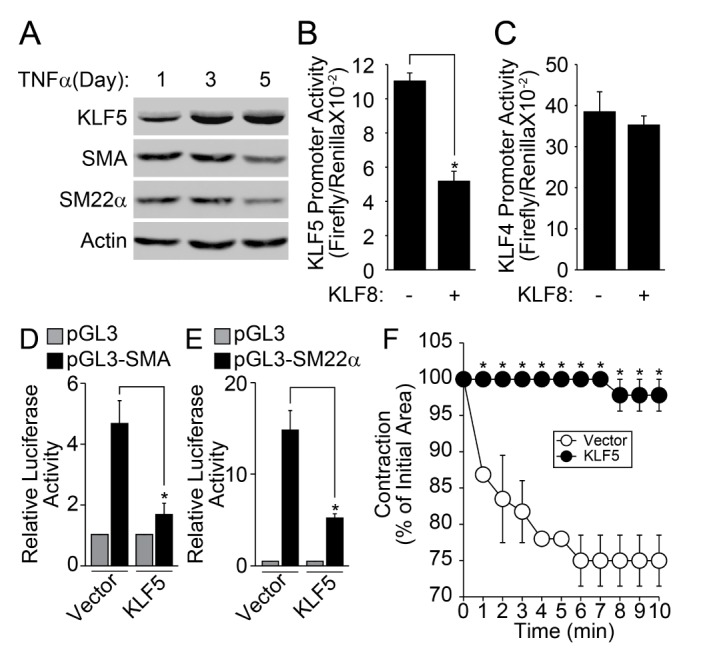
It has been reported that KLF5 plays an essential role in the change of TNFα-induced phenotype of VSMC [22]. Therefore, we examined the effect of KLF8 on the promoter activity of KLF5. The stimulation of contractile type of VSMCs with TNFα markedly enhanced the expression of KLF5, conversely, the expression of VSMC marker genes was down-regulated (Fig. 4A). The co-transfection of KLF8 significantly suppressed the promoter activity of KLF5 (Fig. 4B), whereas the promoter activity of KLF4 was not affected (Fig. 4C). In addition, the co-transfection of KLF5 significantly reduced the promoter activity of both SMA and SM22α (Figs. 4D and 4E). Finally, the overexpression of KLF5 in the contractile phenotype of VSMC abolished the contraction in response to AngII stimulation (Fig. 4F).
DISCUSSION
The modulation of VSMC plasticity is often found in a variety of vessel-associated diseases, such as atherosclerosis, arteriosclerosis, and peripheral blood vessel diseases [1]. Blood vessel integrity and architecture are destroyed by inappropriate proliferation and migration of VSMC, which results in loss of vessel contractility and occlusion [1]. These pathological responses are mainly caused by a phenotypic conversion of VSMC [1]. Therefore, unveiling the mechanistic pathway fundamental to phenotypic conversion of VSMC would provide useful insight into the pathogenesis of vessel-associated diseases.
Several key environmental cues that modulate VSMC plasticity have been reported. For example, the contractile phenotype of VSMC is markedly converted into the synthetic phenotype by the simulation with platelet-derived growth factor (PDGF) [21]. On the other hand, insulin, insulin-like growth factor-1 (IGF-1), and laminin-mediated integrin signaling pathways stimulate the phenotypic switch, from the synthetic type to the contractile type [202324]. In the disease status, it has been reported that the inflammatory response is closely related with the phenotypic conversion of VSMC. For instance, the loss of TNFα significantly suppresses VSMC proliferation, reducing the wall thickness and lesion size during the developmental stages of atherosclerosis [1819]. Likewise, our results show that the stimulation of contractile type of VSMCs with TNFα and PDGF downregulated the marker gene expression, such as SMA, calponin, and SM22α (Fig. 1A). Therefore, inflammatory cytokines may provide key environmental cues for the phenotypic change of VSMC during the pathogenesis of cardiovascular diseases.
The differentiation or dedifferentiation of VSMC is controlled by a coordinated regulation of transcriptional factors [3]. In the present study, we provide evidence that transcriptional activity of myocardin, KLF4, KLF8, and KLF5 is cross-regulated to orchestrate the control mechanism of the VSMC phenotype. KLF8 has been identified as a transcriptional repressor that suppresses target gene expression in association with c-terminal binding protein (CtBP), which is also known as transcriptional repressor [25]. Although the function of KLF4 and KLF5 in the physiology of VSMC has been well established [2627282930], the role of KLF8 in VSMC is still ambiguous. In the present study, we have provided evidence that KLF8 stimulates the differentiation of VSMC. For example, the expression level of KLF8 was markedly reduced during the TNFα-induced dedifferentiation of VSMC (Figs. 1B and 1C). In addition, the expression of KLF8 was highly elevated in the contractile phenotype of VSMC compared with the synthetic phenotype, and forced expression of KLF8 in the synthetic phenotype of VSMC significantly enhanced the expression of contractile marker proteins (Fig. 3). Finally, the ectopic expression of KLF8 significantly repressed the promoter activity of KLF5, which plays an important role in TNFα-induced dedifferentiation of VSMC [22]. Therefore, we can suggest that KLF8 plays an essential role in the differentiation of VSMC.
Furthermore, we raised an important issue regarding the determination of VSMC phenotype. Although each transcriptional regulator seems to act independently during the differentiation or dedifferentiation process, we observed that each transcriptional regulator cross-regulates the transcriptional activity of one another. Previously, it has been reported that myocardin is associated with serum response factor (SRF) transcription factor to directly regulate the expression of smooth muscle marker genes [3132]. In addition to this direct effect on SMC marker gene promoter, our results show that myocardin activated the promoter activity of KLF8 (Fig. 2C). The activation of KLF8 promoter activity by myocardin seems not to be mediated by direct action on KLF8 promoter region, but rather enhanced the promoter activity of KLF4 (Fig. 2D), which in turn up-regulated the promoter activity of KLF8 (Fig. 2E). It is noteworthy that the promoter region of KLF8 possesses the binding sites of KLF4 (Fig. 2A). In addition, it has been reported that KLF4 stimulates the differentiation of VSMC [33]. The promoter region of KLF8 also contains the NFκB binding site (Fig. 2A). It has previously been reported that the transcriptional activity of NFκB is regulated by the PI3k/Akt signaling pathways [343536]. Accordingly, our results show that the modulation of Akt1 expression significantly affected the promoter activity of KLF8 (Figs. 2F and 2G). It also has been reported that FAK-dependent gene expression of KLF8 is mediated by the PI3K/Akt signaling pathway in human ovarian epithelial cancer cells [37]. Furthermore, it has been reported that Akt1 regulates the differentiation of VSMC [20]. As such, the expression of KLF8 is regulated by multiple transcriptional regulators, such as myocardin, KLF4, and NFκB. Therefore, it is reasonable to suggest that various signals merge with the expression of KLF8, and its expression plays an essential role in the regulation of VSMC phenotype.
Currently, the mechanism underlying KLF8-induced VSMC differentiation is still ambiguous. One possible mechanism may be the KLF8-dependent suppression of KLF5. Our results show that TNFα downregulated the expression of KLF8, whereas the expression of KLF5 was significantly enhanced by TNFα (Figs. 1B, 1C, 4A). Moreover, the ectopic expression of KLF8 suppressed the promoter activity of KLF5 (Fig. 4B). The forced expression of KLF5 suppressed the expression of contractile marker genes, such as SMA and SM22α (Figs. 4D and 4E), leading to the loss of AngII-induced contraction of VSMC (Fig. 4F). Congruently, previous reports have shown that the expression of KLF5 is significantly elevated in proliferating VSMC [2938]. It also has been reported that the expression of KLF5 is suppressed in contractile phenotype of VSMC but is reactivated by various environmental cues, such as atherosclerosis, vein graft, and vascular injury [394041]. In these regards, KLF8-dependent regulation of KLF5 expression may very well play an essential role in the phenotypic conversion of VSMC.
In conclusion, the expression of KLF8 is mediated by myocardin, KLF4, and NFκB signaling pathways, thereby stimulating the differentiation of VSMC. KLF8 blocks the expression of KLF5 which plays an essential role in the dedifferentiation of contractile VSMC. In the presence of TNFα, which is an important inflammatory cytokine, the expression of KLF8 is downregulated, thereby relieving the repression activity of KLF5, which leads to the expression of KLF5. Consequently, KLF5 induces the phenotypic conversion of VSMCs and stimulates the loss of contractile function of smooth muscles. Therefore, KLF8 could be the potential therapeutic target for vascular diseases.
 XML Download
XML Download