

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive and ultimately lethal disease characterized by an increase in pulmonary arterial pressure (>25 mmHg) resulting from the thickening of arterial walls and eventually leading to vascular remodeling and right ventricular hypertrophy [12]. Regarding the pathogenic mechanism of PAH, abnormal pulmonary arterial smooth muscle cell proliferation and potential endothelial damage play crucial roles [3456]; however, vascular functions and their underlying molecular mechanisms remain largely unknown.
PAH is known as an idiopathic disorder and can be related to secondary causes such as familial, infectious, and other medical pathologic conditions [137]. Over the past three decades, numerous studies have resulted in major advances in pharmacotherapy of PAH by using various animal models [3891011]. In order to explore molecular mechanisms and evaluate potential therapeutic approaches to PAH, two animal models have been used most commonly, monocrotaline-induced PAH (PAH-MCT) and chronic hypoxia-induced PAH (PAH-CH). In particular, the PAH-MCT rat model has been continuously used in many studies of PAH because the technical procedure for producing this animal model is very simple, inexpensive, and reproducible [121314].
Hypoxic pulmonary vasoconstriction (HPV) is a physiological compensatory phenomenon preventing the ventilation/perfusion mismatch in the lung. Previous studies have shown that the exposure to CH for several weeks attenuates HPV responses and induces molecular changes such as the inhibition of Rho-kinase, downregulation of oxygen-sensitive ion channels, alteration in the mitochondrial reactive oxygen species generation, nitric oxide production, among others [101516171819]. Although PAH-MCT has frequently been used as a model of PAH, the vascular functions, including HPV, have rarely been investigated in comparison with the PAH-CH model. Based on this background, here we investigated and compared the changes in HPV and vascular reactivity by using isolated ventilated/perfused lungs (V/P lungs) from PAH-MCT and PAH-CH rats.
METHODS
Animals
All animal care and experimental procedures were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (IACUC approval no.: 111129-1-1). Male Sprague–Dawley rats (230~280 g) were used for all treatments. MCT-induced PAH was developed by a single intraperitoneal (i.p.) injection of MCT (60 mg/kg) (Sigma, St. Louis, MO, USA), and the rats were sacrificed 21 days later. For CH-induced PAH, rats were exposed to a normobaric hypoxia chamber (21 days of 10% pO2) with an automatic oxygen controller (ProOx Model 110, Biospherix, USA). In the hypoxic chamber, a CO2 absorbent (W.R. Grace, USA) was added to protect from hypercapnia.
Measurement of pulmonary arterial pressure and HPV in isolated ventilated/perfused lungs (V/P lungs)
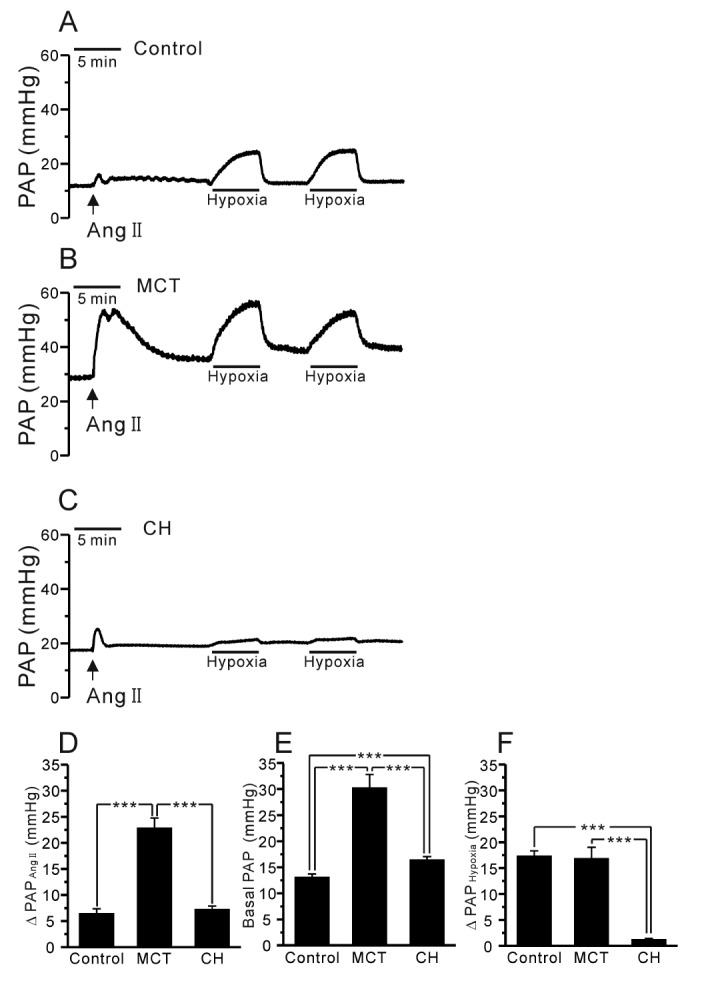
The rats were fully anesthetized with pentobarbital sodium (100 mg/kg, i.p. injection). To confirm adequate anesthesia, the pedal withdrawal and palpebral reflex were tested before the experiment. Tracheostomy was performed, and then the rats were ventilated with an inspired gas mixture (21% O2, 5% CO2, and 74% N2) via a rodent ventilator (respirator 645, Harvard Apparatus, USA). The tidal volume (10 mL/kg) and respiratory rate (85 breaths/min) were continuously maintained. After performing a median sternotomy and heparin injection into the right ventricle (RV), a cannula was inserted into the pulmonary artery (PA), and the suture was tightened. To maintain the constant perfusion of the blood flow, another cannula was inserted into the left atrium via left ventriculotomy. The perfusate consisted of 20 mL of whole blood, and 30 mL of a physiological salt solution (PSS) was added via a peristaltic pump (Servo amplifier 2990, Harvard Apparatus, USA). The flow rate of the perfusate was set at 15 mL/min at rest. The mean pulmonary arterial pressure (PAP) was continuously monitored with a pressure transducer (Abbott Laboratories, USA), and the data were recorded using the Powerlab/4ST and Chart5 software (AD Instruments, Australia). After achieving a stable basal PAP, repetitive HPV was obtained with a hypoxic gas mixture (3% O2, 5% CO2, and 92% N2) in 5 min. As a preconstrictor, angiotensin II (Ang II, 1 µg) was applied, and the perfusate temperature was maintained at 37℃.
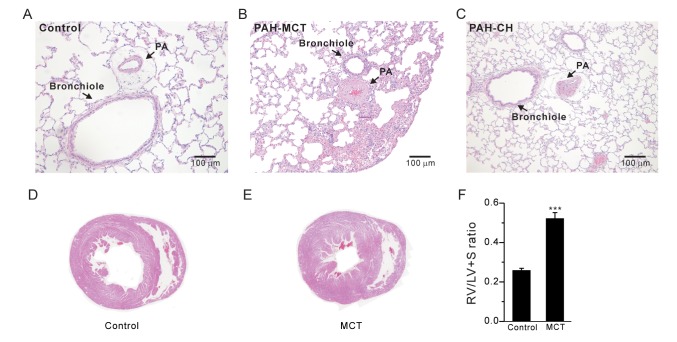
Histology of pulmonary arterial hypertension and right ventricular hypertrophy
Lung tissues were perfused with phosphate-buffered saline (PBS) before fixation. Lung and heart tissues were fixed in 4% paraformaldehyde overnight. For histological analysis, paraffin-embedded tissue sections were cut at 5 mm and stained with hematoxylin and eosin (H&E). Digital images of stained tissues were obtained at 200× magnification using Olympus BX51 and the Adobe photoshop 7.0 software. The right ventricular hypertrophy index was evaluated as a ratio of the RV weight versus the summed weight of the left ventricle (LV) and septum (S) as follows: RV/(LV+S).
Solutions and drugs
For the isolated ventilated/perfused lungs, the PSS consisted of the following (in mM): NaCl 131, KCl 4.7, MgSO4 1.17, NaHCO3 22.61, KH2PO4 1.18, Glucose 10, CaCl2 3.2, sodium meclofenamate 0.0050 g/L, and bovine serum albumin (BSA) 30.0 g/L. The pH was adjusted with 5% CO2 gas. A 40 mM KCl-PSS was prepared by equimolar substitution of NaCl in the PSS. All drugs and chemicals, including BSA, were obtained from Sigma.
Statistical analysis
Data are shown as original recordings and bar graphs of the mean±standard error of the mean. Unpaired Student's t-test or one-way analysis of variance (ANOVA) was used for statistical analysis. Statistical significance was defined as a p-value less than 0.05, 0.01 and 0.001 (*p<0.05, **p<0.01, ***p<0.001).
RESULTS
The index of RV hypertrophy, RV/(LV+S), significantly increased in the PAH-MCT rats compared to that in the control rats (Fig. 1F, n=7; control, n=11; PAH-MCT). Indeed, histological analysis of the PAH-MCT lungs and hearts confirmed the RV hypertrophy and medial thickening of the PA (Fig. 1A~E). These results were similar to those obtained for the PAH-CH rats (Fig. 1C, n=13).
In the V/P lungs from the PAH-MCT and PAH-CH rats, the basal PAP was significantly higher than that in the lungs from the control rats (Fig. 2A~C, E, n=7; control, n=11; PAH-MCT, n=13; PAH-CH). Interestingly, the amplitude of HPV, an increase of PAP by acute hypoxia (3% Po2, 5 min), was similar between the PAH-MCT and control rats, whereas HPV almost disappeared in the PAH-CH rats (Fig. 2A~C, F). In each experiment of V/P lungs, Ang II was initially applied as a pre-condition for the proper HPV responses. Interestingly, the transient PAP increase induced by Ang II was several fold higher in the PAH-MCT rats than in the control of PAH-CH rats (Fig. 2D).
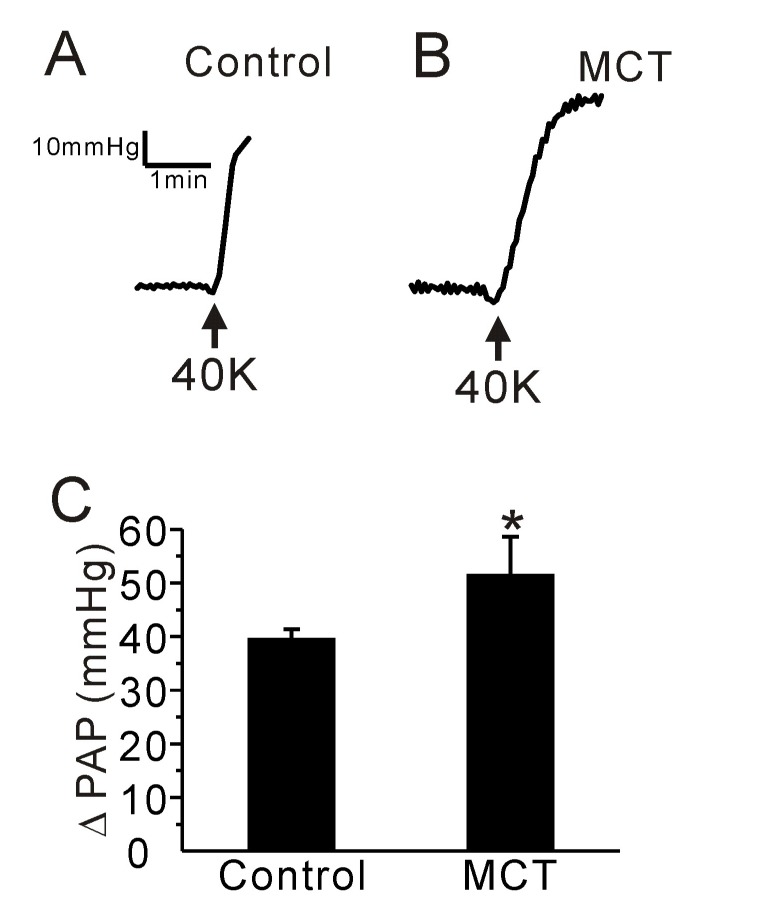
We also compared the PAP increase (ΔPAP) induced by membrane depolarization of PA smooth muscle. High potassium (40 mM KCl)-induced ΔPAP was higher in PAH-MCT than control rats (Fig. 3A~C, n=6, respectively).
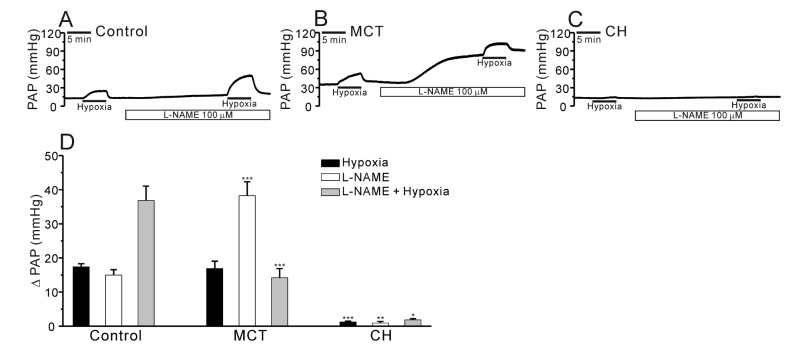
Previous studies have reported that MCT causes endothelial cell dysfunction and decreases nitric oxide (NO) generation in the PA [142021]. To test whether the contribution of endogenously produced NO to the PAP in V/P lungs, we applied NG-nitro-L-arginine methyl ester (L-NAME), a nitric oxide synthase (NOS) inhibitor, to the perfusate. The incubation with L-NAME for 20 min increased the basal PAP in both control and PAH-MCT while the L-NAME effects were much higher in PAH-MCT. Interestingly, the HPV in the presence of L-NAME was less prominent in PAH-MCT. Also it was notable that neither the basal PAP nor the HPV of PAH-CH was affected by L-NAME treatment (Fig. 4A~D, n=6; control, n=8; PAH-MCT, n=5; PAH-CH).
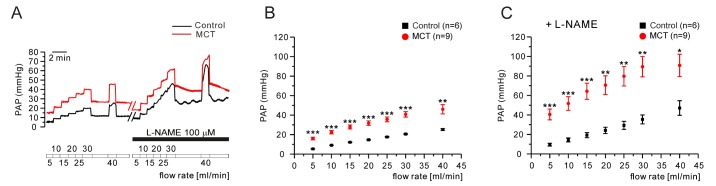
Next, we tested whether a forced increases in the flow rate of V/P lungs show different responses of PAP changes between control and PAH-MCT. The tested range of the flow rate was up to 45 mL/min, and the successive increment was 5 mL/min. In the PAH-MCT rats, the PAP levels under the increased flow rates were consistently higher than those in the control rats (Fig. 5A, B n=6;control, n=9;MCT). Interestingly, in the presence of L-NAME, the PAP increase along with the increased flow rate became steeper in the PAH-MCT and control rats (Fig. 5A, C).
DISCUSSION
In the present study, we investigate the HPV in two different animal models of PAH; PAH-MCT and PAH-CH rats. Although the medial thickening of PA appeared similar between the two models, the responses to Ang II and acute hypoxia showed significant differences. In particular, HPV was still intact in the PAH-MCT rats, while it almost disappeared in the PAH-CH rats.
According to previous studies, the attenuation or loss of HPV has been consistently observed in PAH rat models exposed to chronic hypoxia for 3~4 weeks [22232425]. In PAH-MCT animals, however, the preservation of HPV has been briefly discussed in only two previous studies, without referring to underlying mechanisms [1326]. In this study, here we clearly demonstrate vigorous HPV in the rats three weeks after MCT injection. Compared to the control, the amplitude of the PAP increase upon acute hypoxia exposure was similar in the PAH-MCT rats. However, it has to be noted that the basal PAP was about 10 mmHg higher than that in the control demonstrating the pulmonary hypertension.
Previous studies have shown that the patterns of vascular remodeling were different between PAH-MCT and PAH-CH models [112728]. Neointimal proliferation and medial hypertrophy were not as severe in PAH-MCT as they were in PAH-CH [11]. Besides, vascular changes in PAH-MCT differed among PAs of different diameters [27]. Although we have not rigorously investigated the histological differences in the present study, our study provides an intriguing and noticeable functional differences between the two models of PAH.
Regarding the disappearance of HPV in a relatively early period of chronic hypoxia, our previous study demonstrated that functional downregulation of an oxygen- and thromboxane A2-dependent nonselective cation channels could be involved [1725]. Similar electrophysiological investigation of HPV-related ion channels in the PA smooth muscle cells of the PAH-MCT is requested in future.
In addition to the difference in HPV, our present study demonstrate another contrasting response to NOS inhibitor; the application of L-NAME induced a huge increase of basal PAP in PAH-MCT whereas on a mild or no responses in control and PAH-CH, respectively (Fig. 4B, D). The amplitude of HPV was augmented by L-NAME treatment in the control while not in PAH-MCT. In PAH-CH, HPV was still not observed in the presence of L-NAME (Fig. 4A~D). Since the amplitude of HPV was not augmented in PAH-MCT, we cautiously suggest that an attenuated vasorelaxing influence from eNOS activity as well as a putative increase of endogenous vasoconstrictors might underlie, at least partly, in the enhanced basal PAP and HPV in PAH-MCT rats.
As for the pathophysiological mechanism of PAH-MCT, it is known that MCT metabolites firstly induce pulmonary endothelial cell damage, which results in downregulation of endothelial NOS (eNOS) in the PA [21]. Such inhibition of NOS increased the PA tone and PAP due to the uncompensated actions of intrinsic vasoconstrictors such as endothelin. The downregulation of eNOS activity might show reginal differences and irregular vasoreactivity in the pulmonary circulation [1326].
With the decreased inner diameters of PAs in the PAH-MCT rats, it is suspected that the speed of blood flow and shear stress in each PA branches would be increased. Under the increased mechanical stimuli to the PA endothelium, NO may become a more important factor regulating the PA tone [293031]. Therefore, we tested the effects of a stepwise ascending flow rate in the PAH-MCT and control rats in the presence/absence of L-NAME. Interestingly, the slope of PAP increase according to the ascending flow rate was not significantly different between control and the PAH-MCT rats (Fig. 5A, B). The sensitivity of PAP increase to the increased flow rate was strengthened in the presence of L-NAME (Fig. 5A, C). These results support those of some previous studies suggesting that endogenous NO plays a crucial role in maintaining the basal PAP against an enhanced vasoconstrictor response in PAH-MCT [32]. Increased vasoconstrictors in PAH-MCT would balance the effects of endogenous NO as opposite actions.
Our study also shows a stark increase of Ang II-induced PAP increase in PAH-MCT rats. It is well known that Ang II binds to two Ang II receptor types, type 1 (AT1) and type 2 (AT2). Depending on the receptor type, Ang II has a dual effect on the regulation of vascular contractility. In the PA, Ang II mainly plays a role as a potent vasoconstrictor that binds to the AT1 receptor and is therefore widely used as a preconstrictor inducing HPV in isolated-lung models. Increased AT1 receptor activity was observed in patients with idiopathic PAH [33]; however, the relationship between AT1 expression and vascular remodeling of PAH in animal models remains unclear. In our study, the augmented effect of Ang II in the PAH-MCT rats may suggest the possibility of enhanced AT1 receptor expression.
In summary, this study compared HPV and vascular contractility between PAH-MCT and PAH-CH rats. While HPV was almost abolished in the PAH-CH model, intact HPV was observed in the PAH-MCT rats. In addition, augmented effects were shown by vasoconstrictors such as Ang II and NOS inhibitor in the PAH-MCT rats. These results suggest that PAH-MCT could be an informative animal model for identifying the underlying mechanism of PAH. Further investigation is required to evaluate the effects of various vasoconstrictors in PAH-MCT.
 XML Download
XML Download