

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatocellular Carcinoma (HCC) is the most common type of liver cancer and from a long time, HCC is sixth most common neoplasm and the third most frequent cause of cancer death [1]. Surgical resection has been considered the optimal treatment approach, but only a small proportion of patients qualify for surgery. Furthermore, chemotherapy and radiation therapy are largely ineffective, and metastatic diseases frequently develop even after potentially curative surgery [234]. In this context, new therapeutic options for treating HCC must be developed.
Oleanolic acid has been reported to have numerous pharmacological activities, including anti-inflammatory [5], anti-HIV [6], anti-osteoporosis [7] and hepatoprotective effect [89]. Several recent studies have indicated that OA and its derivatives inhibit the growth of cancer cells through cell cycle arrest and the stimulation of apoptosis [10111213]. An important implication of these finding is that this agent might play a useful role in the treatment of cancer. However, little is known regarding the antitumor effects of OA as well as its underlying mechanisms on Hepatocellular Carcinoma.
Autophagy is characterized by accumulation of cytoplasmic double-membraned autophagic vacuoles called autophagosomes [1415]. These double-membrane autophagosomes then fuse with the lysosome vacuole for degradation and recycling macromolecules [16]. Although apoptosis (type I programmed cell death) is the primary mechanism of anticancer-drugs-induced cell death, an alternative cell death pathway termed autophagic cell death (type II programmed cell death) has emerged as an important mechanism of cancer cell death induced by chemotherapeutic agents and is becoming an attractive approach for anticancer therapies [1718].
In the present study, we report a novel autophagic cell death induced by OA in HepG2 cells. Examination of the signaling pathways indicated that this OA-induced autophagic cell death was subject to negative regulation of PI3K/Akt/mTOR or positive regulation of ROS-dependent pathway. Our findings may provide novel insights into the mechanisms underlying the anticancer effects of OA against human hepatocellular carcinoma.
Go to : 
METHODS
Reagents
Oleanolic acid, DCFH-DA and MDC were bought from Sigma Aldrich (St Louis, US). 3-[4, 5-dimehyl-2-thiazolyl]-2, 5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Amresco. Culture Medium (DMEM) and Fetal bovine serum was obtained from Life Technology. Antibodies against LC3, p-mTOR, p-Akt, p-PI3K, p-4EBP1, p-RPS6KP1, P62 were from Cell Signaling Technology (Boston, US). ACTIN from Santa Cruz Biotechnology (Santa Cruz, US). All the other chemicals were of high purity from commercial sources.
Cell culture
Human hepatocellular carcinoma cells HepG2 and SMC7721 were obtained from ATCC. Cells were maintained in DMEM medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, antibiotics (100 U/ml penicillin and 100 U/ml streptomycin), at 37℃ in a humidified atmosphere of 5% CO2.
Cell viability assay
The cells were plated at a density of 1×103 viable cells per well in 96-well plates. Various concentrations of compound were used to treat cells. After incubation for the indicated time, MTT assay was performed to measure cell viability by a 96-well plate reader (Biotech).
Western blot analysis
Cells after treatment were collected and washed by PBS, then suspended by lysate buffer (30 mM Tris, pH 7.5, 150 mM sodium chloride, 1 mM phenylmethylsul-fonyl fluoride, 1 mM sodium orthovanadate, 1% Nonidet P-40, 10% glycerol, and phosphatase and protease inhibitors) for 30 min on ice. After 10,000 g centrifugation for 10 min, the protein content of the supernatant was determined by a BCA protein assay Kit (Pierce, Rochford, IL). The protein lysates were separated by 10% SDS-PAGE and subsequently electrotransferred onto a polyvinylidene diuoride membrane (Millipore Corp., Bedford, MA). The membrane was blocked with 5% nonfat milk for 1 h at room temperature. The blocked membrane was incubated with the indicated antibodies. Protein bands were visualized using Western blotting detection system according to the manufacturer's instructions.
MDC staining
Autophagic vacuoles were labeled with MDC by incubating HepG2 cells grown on coverslips with 0.05 mM MDC in PBS at 37℃ for 10 min. After incubation, cells were washed four times with PBS and immediately observed by fluorescence microscopy (IX71, Olympus).
GFP-LC3 plasmid transfection
HepG2 cells were transfected with 1 µg of GFP-LC3 expressing plasmid in each well of 6-well plates using liptofectamine as per the manufacturer's instructions (Invitrogen). After 4 h, cells were treated with OA, the fluorescence of GFP or GFP-LC3 was viewed under fluorescent microscope (IX71, Olympus).
Measurement of ROS
Cells were cultured in a 6-well plate, and treated with various concentrations of OA. Then the cells were harvested and incubated with DCFH-DA at 37℃ for 20 min and washed twice with cold PBS. DCF fluorescence distribution was detected by flow cytrometry on a FACScan (Becton Dickinson) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Data were analyzed by Cell Quest software.
Statistical analysis
All data represent as means±SEM of three independent experiments. Statistical analyses were performed using one-way analysis of variance (ANOVA), followed by Student two-tailed t test. The differences between groups were considered to be statistically significant when p<0.05.
Go to :
RESULTS
Inhibitory effect of oleanolic acid on proliferation of cancer cells
To evaluate the effect of the OA on cell proliferation of hepatocellular carcinoma cells, we treated HepG2 and SMC7721 cells with various concentrations OA for 24 and 48 hours, and then MTT assay was performed. Results showed that OA inhibited HepG2 and SMC7721 cell proliferation in not only a dose-dependent manner, but also a time-dependent way (Fig. 1).
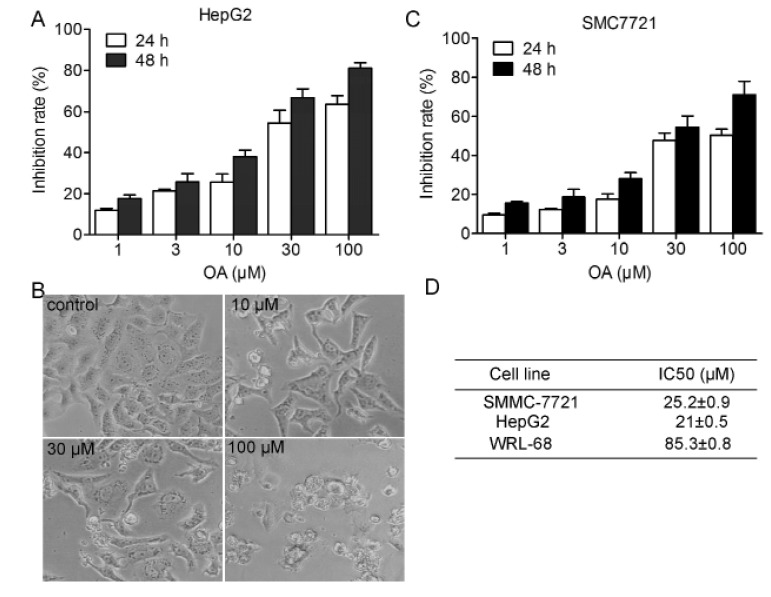 | Fig. 1Dose and time-dependent antitumor effect of oleanolic acid.HepG2 cells were exposed to compound (0~100 µM). Cell proliferation was determined by MTT assay (A) and morphology (B) were observed. (D) Smc7721 cells were exposed to compound (0~100 µm. Cell proliferation was determined by MTT assay. Each column represents the mean of the data from three inde pendent experiments.
|
Oleanolic acid treatment resulted in increased autophagy in HepG2 and SMC7721 cells
Next we were interested to identify whether autophagic cell death was employed when cells were treated with OA. Monodansylcadaverine (MDC) is a specific marker for autolysosomes that localized on the autophagic vacuole membrane structures in the cytoplasm [19]. We examined the distributions of MDC in cells after OA treatment, and found that cells treated with OA showed an increase of MDC dots, indicating the increasing formation of the autophagic vacuoles in comparison with untreated cells (Fig. 2A). The production of LC3-II, which is a cleaved LC3-phosphatidyl- ethanolamine conjugate is another general autophagosomal marker [20]. Conversion of LC3-I to LC3-II significantly increased with the rising dose of OA over 24 h in HepG2 cells and SMC7721 (Fig. 2C and 2E). The LC3-conversion increase also displayed a time-dependent manner (Fig. 2D). Then we used green fluorescent protein (GFP)-fused LC3 to detect autophagy. As shown in Fig. 2B, the formation of GFP-LC3-labeled vacuoles in HepG2 cells was markedly increased 12 h after treatment with 100 µM OA. Taken together, the above findings suggest that OA treatment significantly induces autophagy in HepG2 and SMC7721 cells.
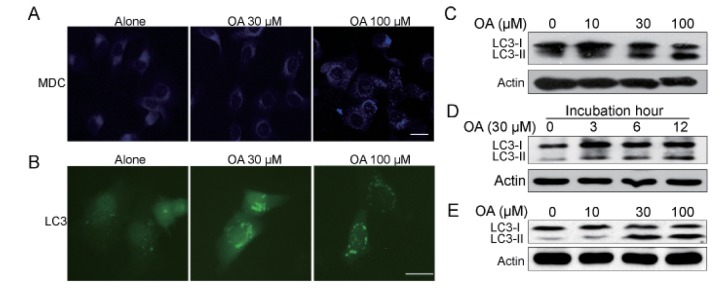 | Fig. 2Oleanolic acid induced evident autophagy in HepG2 cells.(A) HepG2 cells were treated with 100 µM OA for 12 h and then incubated with 0.05 mM monodansylcadaverine (MDC) for 10 min. Cells were then analyzed by fluorescence microscopy. Scale bar: 5 µm. (B) HepG2 cells transfected with GFP-LC3 cDNA were treated with 100 µM OA for 12 h. The formation of vacuoles containing GFP-LC3 (dots) was examined by fluorescence microscopy. Scale bar: 5 µm. (C, D) Protein expression of LC3 in HepG2 cells. (C) HepG2 cells were cultured with indicated concentrations of OA for 24 h. (D) HepG2 cells were cultured with 30 µM OA for 3 h, 6 h, 12 h. (E) SMC7721 cells were cultured with indicated concentrations of OA for 24 h. The results shown are representative of three different experiments.
|
Increased autophagy is responsible for OA-induced cell death in HepG2 and SMC7721 cells
In order to find out whether autophagy contributed to the cell death induced by OA in HepG2 and SMC7721 cells, we examined whether inhibiting autophagy by autophagy inhibitors could affect cell death. First, we found out that in the presence of the autophagy inhibitor 3-MA,the conversion of LC-I to LC3-II was inhibited (Fig. 3A) and MDC-labeled autophagic vacuoles was decreased (Fig. 3B) which means 3-MA treatment significantly inhibited OA- triggered autophagy. Second, while autophagy was interrupted by 3-MA, OA-induced cell death was remarkably prevented (Fig. 3C and 3D). To further confirm the role of autophagy in OA-induced cell death, we also used autophagolysosome fusion inhibitor chloroquine (CQ) in the present study. CQ treatment also decreased cell death induced by OA (Fig. 3C). These results support autophagy as the one of major mechanisms of the cell death in hepatocellular carcinoma cells treated with OA.
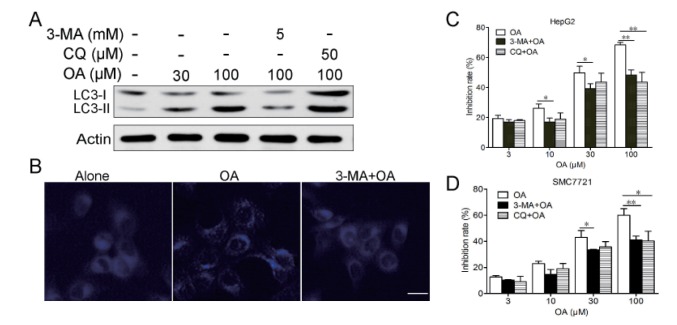 | Fig. 3Autophagy contributed to cell death induced by OA in HepG2 cells.(A) HepG2 cells were treated with 30, 100 µM OA for 12 h in the presence or absence of 5 mM 3-MA or 50 µM CQ, LC3 expression were detected by western blot. (B) HepG2 cells were treated with 100 µM OA for 12 h in the presence or absence of 5 mM 3-MA for 2 h and then stained with MDC. Scale bar: 5 µm. (C) HepG2 cells were treated with various concentrations of OA and/or 1 or 5 mM 3-MA for 24 h. Cell death was measured by MTT assay. (D) SMC7721 cells were treated with various concentrations of OA and/or 1 or 5 mM 3-MA for 24 h. Cell death was measured by MTT assay. *p<0.05, **p<0.01. The results shown are representative of three different experiments.
|
OA induces autophagic cell death through PI3K/AKT1/mTOR and ROS-dependent pathways
During the process of autophagy, p62 acts as an adaptor protein which links LC3 with ubiquitin moieties on misfolded proteins therefore mediated the clearance of these ubiquitylated proteins [21]. In our experiments, we found that expression level of p62 were down regulated by OA treatment in HepG2 cells (Fig. 4). The PI3K/Akt/mTOR signaling pathway is the well-known pathways involved in the regulation of autophagy. So we examined the effect of OA on this pathway by using western blot. After treatment with 10, 30 and 100 µM OA, there was a dose-dependent decrease in the expressions of phosphorylated RPS6KB1, RPS6 (ribosomal protein S6) and 4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) in HepG2 cells (Fig. 4). Furthermore, OA treatment also inhibited the phosphorylation of PI3K and Akt, which are the upstream of mTOR (Fig. 4). Since the reactive oxygen species (ROS) seems to play a central role in autophagy induction [222324], we examined whether ROS was involved in OA-induced autophay process. In order to detect the changes of intracellular ROS, we used flow cytomentry to measure the fluorescence of DCF after OA treatment. As shown in Fig. 5A and 5B, treatment with OA for 12 hours led to an increase of ROS in HepG2 cells in a dose-dependent manner. And with ROS scavenger NAC pretreatment, OA-induced LC3-II production was remarkably inhibited (Fig. 5D). At the same time cell death was significantly reduced (Fig. 5C). These results indicated that ROS was indispensable for OA-induced autophagic cell death.
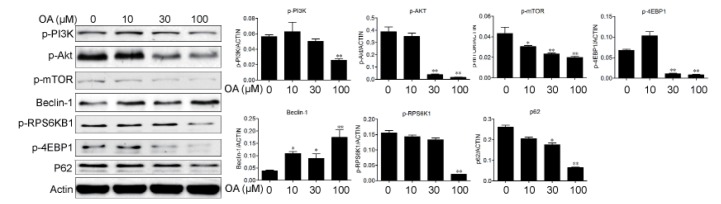 | Fig. 4Oleanolic acid induced autophagy via PI3K/Akt/mTOR pathway.HepG2 cells were cultured with indicated concentrations of OA for 24 h and protein expression were examined in HepG2 cells. The results shown are representative of three different experiments. The band intensities from repeated experiments were calculated and static. *p<0.05, **p<0.01.
|
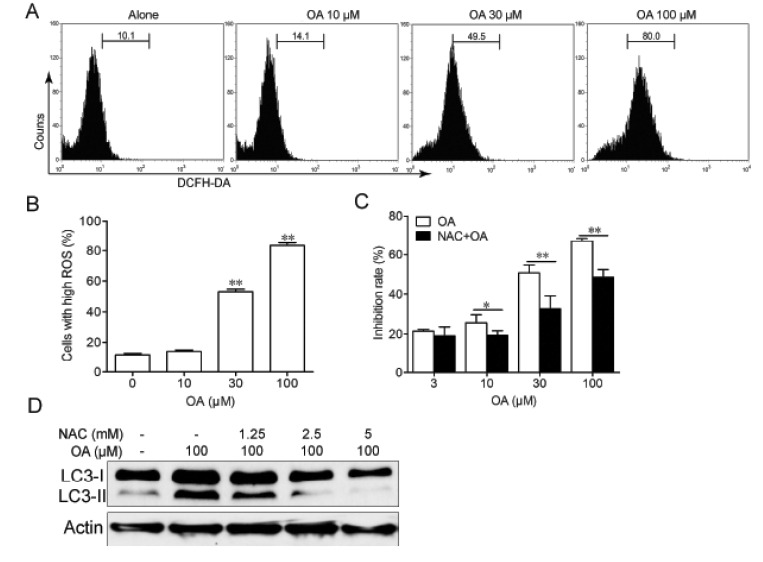 | Fig. 5Autophagy induced by OA in HepG2 cells was ROS-dependent.(A, B) HepG2 cells were treated with indicated concentrations of OA for 12 h and stained with DCFH-DA and analyzed by flow cytrometry. (C) HepG2 cells were treated with indicated concentrations of OA for 24 h in the presence or absence of 5 mM NAC and cell death was measured by MTT assay. *p<0.05, **p<0.01. (D) HepG2 cells were treated with 100 µM OA for 12 h in the presence or absence of indicated concentrations of NAC and expression of LC3 was examined by western blot. The results shown are representative of three different experiments.
|
Go to :
DISCUSSION
In this study, we provided novel evidence that OA activates the autophagic cell death in human hepatocellular carcinoma cells. Our conclusions were based on the following observations: First, we found that punctuated distribution of LC3, elevated ratio of LC3-II to LC3-I, increased p62 degradation and the massive autophagic vacuolization were occurred in OA-treated cells (Fig. 2). Second, OA-induced cell death was mediated through autophagy: the cell death was significantly suppressed by approaches inhibiting autophagy, such as the pretreatment of various autophagy inhibitors (Fig. 3). These results provided a mechanistic characterization of OA-induced cancer cell death.
Autophagy is controlled by mammalian target of rapamycin (mTOR) downstream of phosphatidylinositol3-kinase (PI3K)/Akt, which regulates cell growth and protein synthesis in response to nutrient and growth factor availability [25]. Autophagy is believed to has cytoprotective roles in response to cancer therapeutics. Novel therapies such as the use of proteasome inhibitors [26], or kinase inhibitors [27] could induce autophagy which is believed to blunt drug efficacy. But at the same time, excess autophagy can also act as a pro-death mechanism which lead to the damage of cancer cells. So far, several chemotherapeutic agents such as sodium selenite [28], Arsenic trioxide and Bortezomibhave been reported to be able to induce autophagic cell death [2930]. Previously, OA has been reported to induce apoptotic cell death in cancer cells [31], here we proposed that autophagic cell death, together with apoptosis, both are the mechanisms for OA-induced anticancer activity. Since cancer cells may often develop resistance to apoptosis, autophagy and apoptosis-induction ability of OA may provide an effective option in cancer therapy, especially for apoptosis-resistant cancer.
ROS, including superoxide (O2–), hydroxyl radical (HO–), and hydrogen peroxide (H2O2), are known to be signaling molecules in autophagic cell death. It has been shown that accumulated ROS lead to inactivation of the cysteine protease Atg4, leading to accumulation of the Atg8-phosphoethanolamine precursor that is required for the initiation of autophagosome formation, suggesting ROS can directly activate autophagy [23]. Herein, our results show that OA induced ROS accumulation contributed to autophagic cell death in HepG2 cells in a dose-dependent manner. ROS accumulation can also lead to apoptosis induced by OA and we deduce that OA could induce both apoptotic and autophagic cell death via ROS-dependent pathway.
Taken together, our findings suggest a new autophagic cell death pathway that OA-induced in human hepatocellular carcinoma cells. Our study thus prove OA to be a useful chemical probe for the investigation of autophagy in cancer cells and provide evidence for its being used as an effective chemotherapeutic agent against hepatocellular carcinoma in the clinical settings.
Go to :
 XML Download
XML Download