

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cardiac hypertrophy is an adaptive response to pressure or volume stress, characterized by an increase in the size of individual cardiomyocytes and whole-organ enlargement. Despite this process is initially compensatory, sustained pathologic hypertrophy is deleterious and may lead to heart failure [1,2,3]. Although there has been remarkable development in diagnosis and management of heart failure, the morbidity associated with this ailment is still a major challenge to the economy and public health [4,5]. Current pharmacological approaches to the treatment of heart failure including calcium channel blockers, beta-blockers and renin-angiotensin-aldosterone system inhibitors. In spite of impressive gains with existing pharmacological treatment, patients still have poor quality of life and suffer serious side effects [6,7]. Therefore, the need to explore novel strategies remains. Recently, Epigallocatechin-3-gallate (EGCG), the major component of polyphenols in green tea, has been reported to be cardioprotective in studies using animal models of cardiovascular disease [8]. Moreover, others and our previous study also reported that EGCG could inhibit cardiac hypertrophy and fibrosis both in vivo and in vitro [9,10]. However, the molecular mechanisms underlying the anti-hypertrophic effect of EGCG remain to be clarified, and little is known about the potential target of EGCG on cardiac hypertrophy.
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that can be activated by cellular stresses and ATP depletion [11,12]. It has been shown that activation of AMPK inhibits the development of cardiac hypertrophy via a number of pathways, such as eukaryotic elongation factor-2 (eEF2), p70S6 kinase (p70S6K), and mammalian target of rapamycin (mTOR) [13,14]. Moreover, in AMPK alpha-2 null mice, cardiac hypertrophy induced by isoproterenol or aortic constriction is significantly larger than in controls and is correlated with p70S6K activation [15,16]. Therefore, AMPK is an attractive target to treat or prevent cardiac hypertrophy and subsequent heart failure. Recently, several lines of evidence demonstrated that EGCG could activate AMPK signaling pathway in cancer cells and in mice [17,18]. However, it is not known whether this regulatory effect is related to a protective role cardiac dysfunction. Therefore, the purpose of this study was to examine the effects of EGCG on phenylephrine (PE)-induced cardiac hypertrophy in H9C2 cardiomyocytes, and we further investigated whether the AMPK-dependent pathway is involved.
Go to : 
METHODS
Reagents
EGCG (purity>95%) was purchased from Sigma aldrich. Dulbecco's Modified Eagle Medium (DMEM) and foetal bovine serum (FBS) were purchased from Gibco (Logan, UT, USA) and Sijiqing Biological Engineering Materials Co Ltd. (Hangzhou, China), respectively. Bovine serum albumin (BSA) was purchased from Sangon (Shanghai, China). Antibodies against AMPK, phospho-AMPK (T172) and phospho-p70S6K (T389) were purchased from Cell Signaling (Cell Signaling Technology); anti-β-actin antibody was purchased from Santa Cruz Biotech (Santa Cruz Biotechnology); anti-phospho-eEF2 and total-eEF2 was purchased from bioworlde (Guangzhou, China). EGCG was dissolved in distilled water at 100µM, and stored at -20℃ until dilution before use.
Cultures of H9C2 cardiomyocytes
H9C2 rat cardiomyocytes were cultured in DMEM supplemented with 10% FBS. Cell culture media were changed every 2 days. Before PE stimulation, H9C2 cells were cultured in DMEM containing 1% FBS for serum starvation.
Quantitative real-time polymerase chain reaction (qRTPCR)
Total RNA from tissues or cultured cells was extracted using Trizol reagent (Invitrogen). One microgram of total RNA was reverse transcripted using One-step RT Kit (Takara Biotechnology) and the resulting cDNA was used as a PCR template. The mRNA expression levels were determined using SYBR-Green Quantitative PCR Kit (Takara Biotechnology) by Applied Biosystems 7500 Fast Real-Time PCR system (ABI). All PCRs were done in triplicate. Rat-specific primers (Table 1) for natriuretic peptides type A (Nppa), brain natriuretic polypeptide (BNP) were synthesized by Invitrogen. GAPDH served as an endogenous control.

Western blotting analysis
Western blotting analyses were performed as previously described [19]. Briefly, protein was separated by SDS-PAGE gel electrophoresis, and then transferred to PVDF membranes (Millipore). After blocking with 5% nonfat milk, the membranes were incubated with primary antibodies, followed by incubation with appropriate horseradish peroxidase (HRP)-labeled second antibodies. Immunoreactive bands were detected with the Super-Signal West Pico Chemiluminescent Substrate (Pierce), and molecular band intensity was determined by densitometry.
Measurement of cell surface area
Cardiomyocytes grown in 48-well plates were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, followed by 0.5% Triton-100 treatment for 5 min. After incubated with 0.1% rhodamine-phalloidin for 30 min, the myocytes were washed in PBS for further interaction with DAPI. The images of cardiomyocytes were detected by High Content Screening system (ArrayScanVTI, Thermo Fisher Scientific, Rockford, Illinois, USA), and the cell surface area from randomly selected fields (50 for each group) was determined by the built-in image analysis software.
Statistical analysis
Data are presented as mean±SE. Statistical analyses between two groups were performed by unpaired Student's t-test. Differences among groups were tested by one-way analysis of variance (ANOVA). In all cases, differences were considered statistically significant with p<0.05.
Go to :
RESULTS
EGCG blocks cardiac hypertrophy in H9c2 cardiomyocytes
In H9c2 cardiomyocytes, EGCG could markedly suppress PE-induced Nppa and BNP gene expression as revealed by qRT-PCR analysis (Fig. 1A). In addition, PE stimulation (100µM for 24 h) led to significant increase in cell surface area, which could be attenuated by preincubation with EGCG (25µM) for 60 min and subsequently treated with 100µM PE (in the presence of EGCG) for 24 h (Fig. 1B and C). However, EGCG itself had no effect on the Nppa and BNP mRNA levels, or surface area of cardiomyocytes.
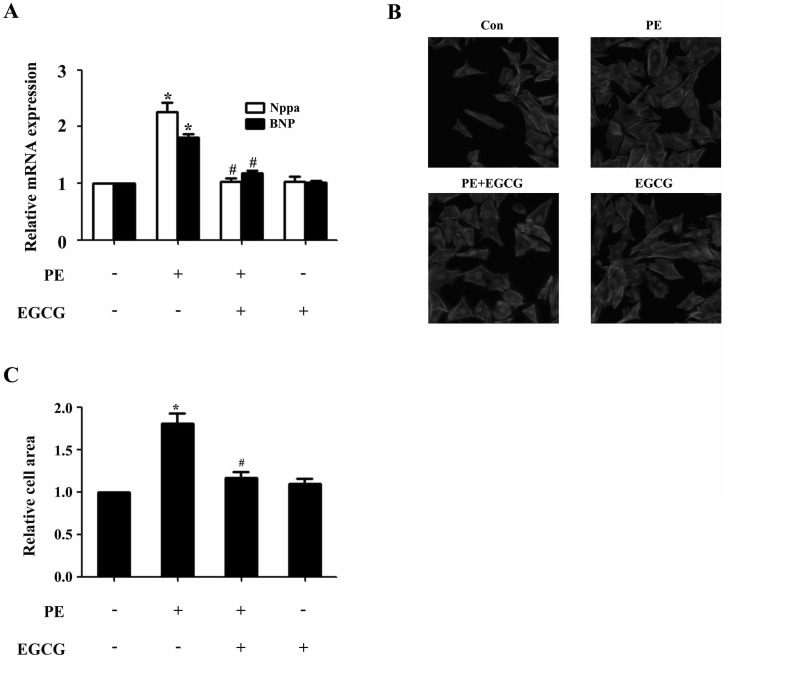 | Fig. 1EGCG blocks cardiac hypertrophy in H9C2 cardiomyocytes. H9C2 cardiomyocytes were pretreated with EGCG (25µM) for 1 h followed by stimulation with 100µM PE for 24 h. (A) Cellular hypertrophy was demonstrated by changes in mRNA levels of hypertrophic biomarkers Nppa and BNP. (B and C) Cardiomyocytes were stained with rhodamine-phalloidin followed by cell surface area quantization. *p<0.05 vs. the control group, #p<0.05 vs. the PE group, n=3.
|
EGCG Treatment increases AMPK phosphorylation and decreases the phosphorylation of p70S6K and eEF2
To evaluate a possible involvement of AMPK, we assessed the phosphorylation status of this key metabolic regulator. Treatment with EGCG led to a induction of AMPK phosphorylation in a time- and dose-dependent manner (Fig. 2A~D). In order to investigate the molecular signaling mechanisms that may contribute to the EGCG-induced reduction in hypertrophic growth, cell lysates from cardiac myocytes treated with PE in the absence or presence of EGCG were subjected to immunoblot analysis using anti-phospho-AMPK antibodies. As shown in Fig. 2E and F, stimulation with 100µM PE for 24 h resulted in downregulation of AMPK phosphorylation, which could be reversed by pretreatment with 25µM EGCG for 60 min. Moreover, although PE increased the phosphorylation status of p70S6K compared with control, EGCG was able to decrease p70S6K phosphorylation to basal levels (Fig. 3A and B). Furthermore, immunoblot analysis of eEF2 using anti-phospho-eEF2 antibody as a marker of its activity (where phosphorylation indicates inactivation) demonstrated that EGCG increased eEF2 phosphorylation in cells in the absence or presence of PE (Fig. 3C and D).
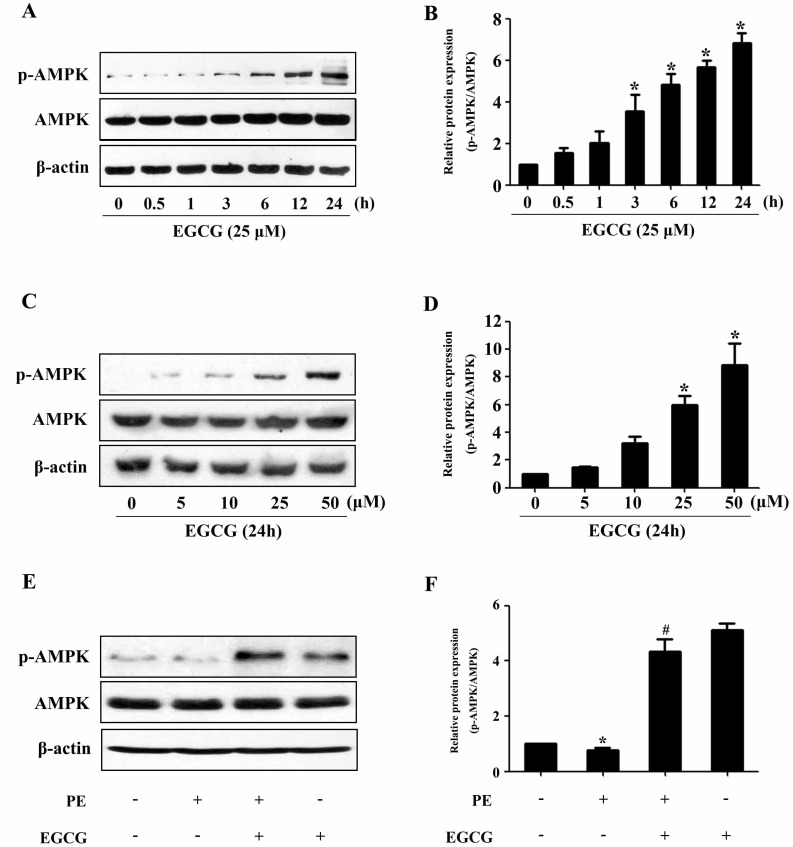 | Fig. 2EGCG treatment increases AMPK phosphorylation. (A and B) H9C2 cardiomyocytes were treated with various concentrations of EGCG for 24 h. The levels of phosphor-/total AMPK were detected. (C and D) Cells were treated with 25µM EGCG for indicated time points and the protein expression of phosphor-/total AMPK were assessed by western blotting. (E and F) Cells were preincubated with 25µM EGCG for 1 h followed by stimulation with 100µM PE for 24 h, and then the Levels of phosphor-/total AMPK were measured by western blotting. *p<0.05 vs. the group without treatment, #p<0.05 vs. the group treated with PE, n=3.
|
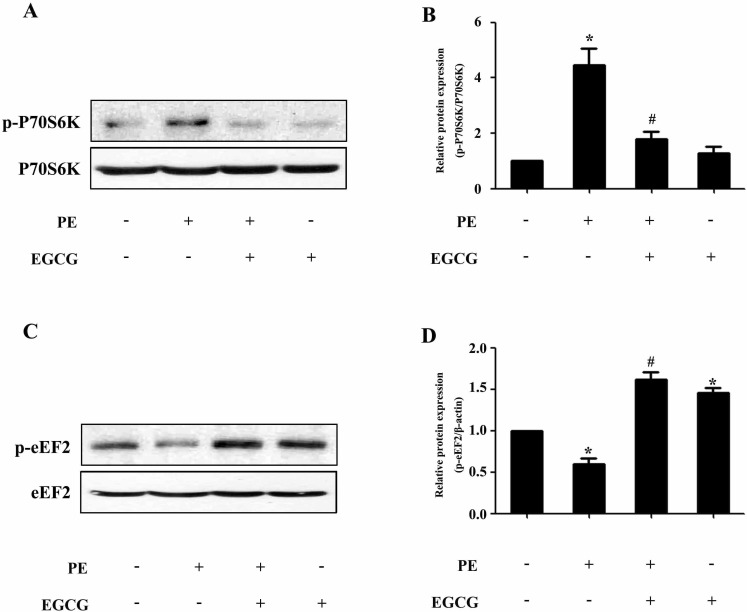 | Fig. 3EGCG decreases the p70S6K phosphorylation and increases the eEF2 phosphorylation. Cells were preincubated with 25µM EGCG for 1 h followed by stimulation with 100µM PE for 24 h. (A and B) The Levels of phospho-/total p70S6K were measured by western blotting. (C and D) The phospho-/total eEF2 were detected by western blotting. *p<0.05 vs. the group without treatment, #p<0.05 vs. the group treated with PE, n=3.
|
AMPK participates in the inhibitory effects of EGCG on cardiac hypertrophy in H9C2 cardiomyocytes
To examine the role of AMPK pathway in the inhibitory effects of EGCG on cardiac hypertrophy, the activity of AMPK in H9C2 cardiomyocytes was blocked by a selective inhibitor (Compound C) prior to the treatment with EGCG. Western blotting analysis showed that the phosphorylation status of AMPK was significantly suppressed after treated with compound C as compared with control (Fig. 4A). Moreover, EGCG failed to inhibit PE-induced increase in the phosphorylation level of p70S6K and EGCG also failed to increase the phosphorylation status of eEF2 inhibited by PE, which suggested involvement of AMPK (Fig. 4E). Then the cell surface area and mRNA levels of Nppa and BNP were measured respectively. As shown in Fig. 4B~D, when AMPK was inhibited by compound C, EGCG failed to inhibit PE-induced increase in cell surface area as well as expression of hypertrophic biomarkers, Nppa and BNP, which suggested involvement of AMPK in the protective effects of EGCG against cardiac hypertrophy.
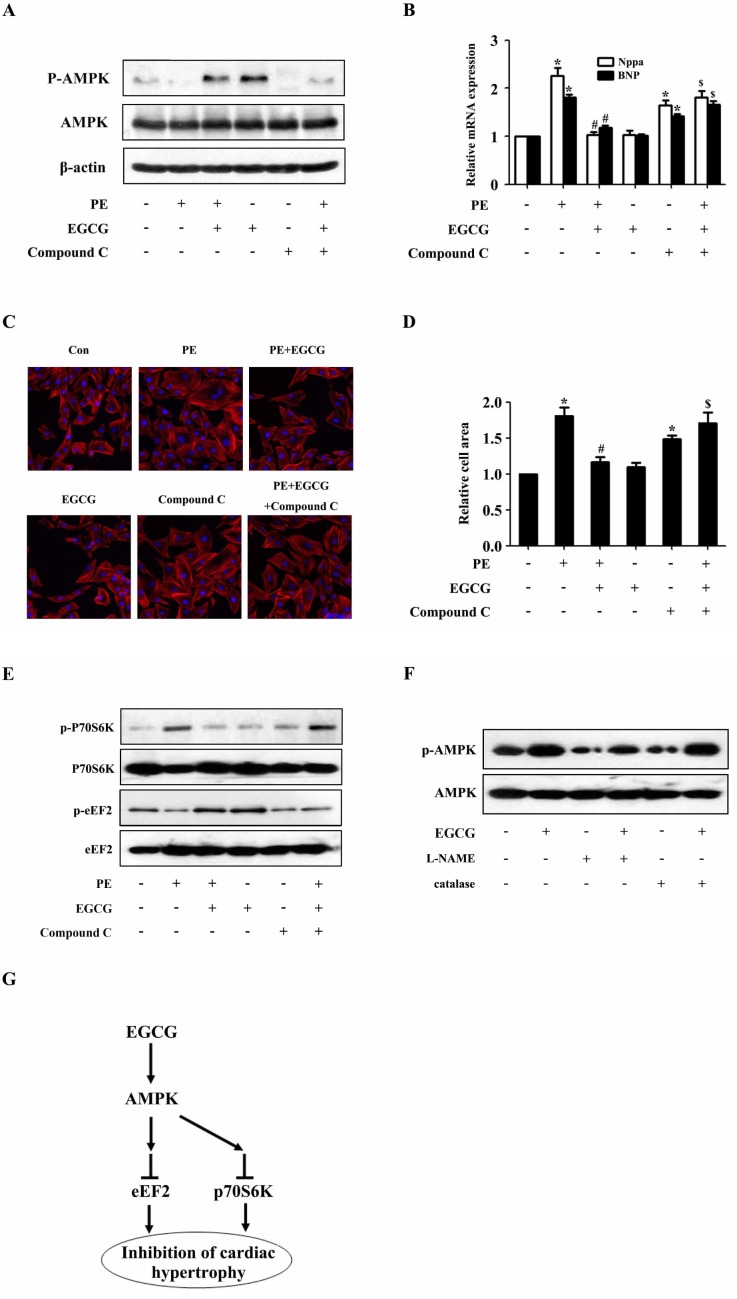 | Fig. 4AMPK participates in the inhibitory effects of EGCG on cardiac hypertrophy in H9C2 cardiomyocytes. H9C2 cardiomyocytes treated with Compound C were pretreated with 25µM EGCG for 1 h followed by stimulation with PE for 24 h. (A) Levels of phospho-/total AMPK were measured by western blotting. (B) Nppa and BNP mRNA levels were measured by quantitative RT-PCR with GAPDH as endogenous control. (C and D) Cardiomyocytes were stained with rhodamine-phalloidin followed by cell surface area quantization. (E) The protein expression of phosphor-/total p70S6K and phosphor-/total eEF2 were detected by western blotting. (F) H9C2 cardiomyocytes were preincubated with a catalase (200 U/ml) or L-NAME (5 mM) for 1 h and subsequently incubated with the indicated concentrations of EGCG for 24 h. Levels of phosphor-/total AMPK were measured by western blotting. (G) Model for EGCG inhibiting cardiac hypertrophy. *p<0.05 vs. the group without treatment, #p<0.05 vs. the group treated with PE alone, and $p<0.05 vs. the group treated with PE plus EGCG, n=3.
|
Role of reactive nitrogen species in EGCG-induced AMPK activation
To investigate whether RNS is involved in the EGCGmediated AMPK regulation in this study, the H9C2 cardiomyocytes were pretreated with L-NAME (a nitric oxide synthase inhibitor) for 1 h followed by EGCG stimulation for 24 h. The protein expression of p-AMPK and AMPK were detected by western blotting. As shown in Fig. 4F, pretreatment with an L-NAME prevented the EGCG enhanced phosphorylation of AMPK. In addition, we showed that treatment of H9C2 cardiomyocytes with catalase did not alter AMPK phosphorylation, indicating that ROS are not involved in EGCG-induced AMPK activation in H9C2 cardiomyocytes.
Go to :
DISCUSSION
Whereas previous studies demonstrated that EGCG is effective in preventing cardiac hypertrophy both in cultured cells and in animal models, the mechanisms involved in the anti-hypertrophic effect have not been clearly defined. Our data showed that EGCG is able to activate AMPK and diminish the activity of both p70S6K and eEF2 in H9C2 cardiomyocytes. Moreover, we further showed that the blockade of AMPK activation prevents EGCG suppression of cardiac hypertrophy, suggesting that EGCG exerts its anti-hypertrophic effects via AMPK activation. These findings shed new light on the mechanism underlying anti-hypertrophic effect of EGCG and support the concept that EGCG could be an effective preventive and therapeutic candidate against cardiac hypertrophy.
EGCG and green tea have been shown to exert remarkable protective effects against the cardiovascular system, including an anti-inflammatory effect [20], anti-oxidative [21] and reducing evolving atherosclerosis [22]. In 2003, Priyadarshi et al. first demonstrated that green tea extract essentially blocked the development of cardiac hypertrophy in the 5/6 nephrectomy rat model [23]. However, the green tea extract which was used in those studies is not a pharmacologically "pure" preparation, but rather a mixture of pigments, theaflavins and thearubigin. Recently, Li et al. showed that EGCG effectively inhibits pressure overloadinduced cardiac hypertrophy in rats and AngII-induced hypertrophy in neonatal rat cardiomyocytes [10]. Consistent with previous study, in this work, we examined the effect of EGCG on PE-induced cardiac hypertrophy in H9C2 cardiomyotytes. The results of the present study showed that EGCG treatment could significantly inhibit the upregulation of hypertrophic related genes and cell surface area stimulated with PE (Fig. 1).
The mechanism by which EGCG mediates its anti-hypertrophic effects remains unclear. AMPK is a critical regulator of energy homeostasis, which stimulates ATP-generating pathways and inhibits ATP-consuming pathways in response to the depletion of ATP by cellular stresses [24,25]. There is increasing evidence for the involvement of AMPK signaling pathway in pathological cardiac hypertrophy and heart failure [26,27,28]. AMPK Activation is believed to decrease protein synthesis induced by PE treatment [13]. Moreover, Activation of AMPK also result in decreased p70S6 kinase phosphorylation and increased phosphorylation of eEF2, suggesting that inhibition of protein synthesis involves the eEF2 kinase/eEF2 axis and/or the p70S6 kinase pathway [29]. Taken together, these results suggest that the inhibition of protein synthesis by pharmacological activation of AMPK may be a key regulatory mechanism by which hypertrophic growth can be controlled. In this study, we showed that AMPK is activated in H9C2 cardiomyocytes by EGCG, and is required for EGCG suppression of cardiac hypertrophy (Fig. 2 and 4). Moreover, we showed that EGCG is able to decrease the p70S6K phosphorylation and increase the eEF2 phosphorylation (Fig. 3). Convergently, it suggests that EGCG may inhibit cardiac hypertrophy via regulating AMPK in H9C2 cardiomyocytes.
Oxidative stress is considered to be a major cause for the development of pathological cardiac hypertrophy [30]. Previous studies demonstrated that ROS and RNS, such as hydrogen peroxide and peroxynitrite, play an important role in the activation of AMPK respectively. ROS could stimulate contraction-mediated activation of glucose transport through AMPK activation [31]. Moreover, it has been reported that EGCG treatment of HT-29 colon cancer cells inhibits cell proliferation and stimulates hydrogen peroxide-mediated phosphorylation of ACC [32], suggesting a role for ROS as an upstream regulator of AMPK. In addition, AMPK activation by metformin, a well-known antidiabetic drug, is reported to be mediated by RNS [33,34]. Further, Thandapilly et al showed that resveratrol prevents norepinephrine induced hypertrophy in adult rat cardiomyocytes, by activating NO-AMPK pathway [35]. However, it is not currently known whether ROS and RNS are involved in the EGCG-mediated AMPK regulation in PE-induced cardiac hypertrophy in H9C2 cardiomyocytes. Therefore, we examined whether EGCG-induced AMPK activation is mediated by ROS or RNS in H9C2 cardiomyocytes. In this paper, we showed that pretreatment with an L-NAME prevented the EGCG enhanced phosphorylation of AMPK, suggesting that RNS are involved in the EGCG-induced activation of AMPK. On the other hand, treatment of the cells with catalase did not alter AMPK phosphorylation, indicating that ROS are not involved in EGCG-induced AMPK activation in H9C2 cardiomyocytes (Fig. 4F). Although we found that RNS were participate in the regulation of EGCG on AMPK pathways, further studies are warranted to elucidate in detail the mechanism underlying EGCG-induced activation of AMPK.
In conclusion, our present work demonstrated that EGCG inhibits cardiac hypertrophy in response to PE in H9C2 cardiomyocytes. EGCG prevented the development of cardiac hypertrophy by activating AMPK-dependent signaling pathway (Fig. 4G). This study is relevant to the understanding of the inhibitory effect of EGCG on cardiac hypertrophy and related molecular mechanisms. It may also provide some useful information for elucidating the signaling pathways leading to cardiac hypertrophy mediated by PE. However, it is currently still unclear how EGCG exactly activates AMPK. Future investigation on this matter may provide new therapeutic approaches for the management of cardiac hypertrophy.
Go to :
 XML Download
XML Download