

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway, which was first identified in 1990s1, has been known to be activated during the early phase of the onset of lung cancer2, thereby causing cell growth, proliferation, angiogenesis, and synthesis of various proteins3,4. If PI3K and Akt are activated by the stimulation of various growth factors, they activate mTOR. The activated mTOR in turn regulates eukaryotic initiation factor 4E binding protein-1 and 40S ribosomal protein S6 kinase (p70S6K) that are involved in the regulation of protein synthesis3,4. It has been reported that PIK3CA gene mutation and its amplification are observed in approximately 2% and 12~17%, respectively, of patients with non-small cell lung cancer5-7. These are associated with increases in PI3K activity and Akt expression.
Several drugs that inhibit PI3K/Akt/mTOR pathway have been currently developed and are under investigation. Temsirolimus and everolimus, mTOR inhibitors, have been already clinically studied in a phase III clinical study conducted on renal cell carcinoma patients, and they have been released into the market8,9. For non-small cell lung cancer, various drugs including temsirolimus and everolimus have been undergoing clinical trials based on their anti-cancer effect identified in experiments using cells10-14.
This study was conducted to compare the effect of the co-administration of temsirolimus, a mTOR inhibitor, and GSK69069315, an Akt inhibitor with that of the sole administration of each drug on cancer cell survival. In addition, changes in apoptosis and autophagy after administration were also investigated.
Materials and Methods
1. Cell culture and reagents
A549 and NCI-H460 lung cancer cell lines were purchased from American Type Culture Collection (ATCC; Rockville, MD, USA). Each cell line was cultured in RPMI1640 medium containing 10% fetal bovine serum and 1% gentamicin sulfate in a CO2 incubator (37℃, 5% CO2). Temsirolimus, a mTOR inhibitor, was purchased from Selleck Chemicals (Houston, TX, USA), and GSK690693, an Akt inhibitor, was provided from GlaxoSmithKline Korea (Seoul, Korea). Methylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide (MTT) and propidium iodide (PI) were purchased from Sigma (St. Louis, MO, USA), and annexin V-FITC was purchased from BD Bioscience (San Jose, CA, USA). Protein assay kit, which can quantify proteins, was purchased from Bio-Rad (Richmond, CA, USA). Antibody to caspase 3, antibody to beclin 1 and secondary antibodies were purchased from Cell Signaling (Boston, MA, USA). Antibodies to poly(ADP-ribose) polymerase (PARP), light chain (LC) 3B, p-PRAS40, p-p70S6K, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Enhanced chemiluminescence (ECL) kit was purchased from PerkinElmer (Waltham, MA, USA).
2. Methylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide (MTT) analysis
6×103 cells were placed into a 96-well plate, and cultured for more than 12 hours. Then, temsirolimus and GSK690693 were added to the cultured cells for 72 hours according to each concentration. MTT reagent was added to each plate. Three hours later, 10% sodium dodecyl sulfate solution was added to dissolve purple formazan which was formed by the live cells. After 24-hour culture, the result was analyzed at 595 nm using a microplate reader (Bio-Rad).
3. Combination index (CI) calculation
For the statistical analysis of the synergistic effect of drug co-administration on MTT analysis, combination index was calculated using CalcuSyn® software version 2.1 (Biosoft, Cambridge, UK). If CI<1, it refers to synergistic effect. If CI=1, it refers to additive effect. If CI>1, it refers to antagonism.
4. Apoptosis assay
4×105 cells were cultured at a 60 mm dish for one day. On the next day, the cultured cells were treated with temsirolimus and GSK690693, followed by cell collection 48 hours later. The cells were placed in annexin V binding buffer (150 mM NaCl, 18 mM CaCl2, 10 nM HEPES, 5 mM KCl, 1 mM MgCl2), and then treated with annexin V (1 g/mL) and 50 g/mL PI, followed by reaction for 30 minutes in a dark place. Then, they underwent fluorescence-activated cell sorting (FACS), and were analyzed using CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA).
In addition, the cells were stained using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit (Roche, Basel, Switzerland), followed by observing apoptotic cells using a confocal laser scanning microscope.
5. Acridine orange staining
For the detection of the acidic pH of autophagolysosomes that appear during autophagy, acridine orange staining was performed. The cells that were treated with drugs were stained 2µg/mL of acridine orange solution for 15 minutes, followed by observing using a confocal laser scanning microscope.
6. Western blot
To examine changes in proteins related to apoptosis or autophagy, the cultured cells were collected and then underwent lysis in lysis buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, 1 mM sodium vanadate, 10 mM sodium pyrophosphate, 10 mM NaF, 300µM p-nitrophenyl phosphate, 1µg/mL leupeptin, 1 mM phenylmethanesulfonylfluoride, 10 µg/mL aprotinin, pH 7.3), followed by centrifugation and quantification. Western blot was conducted in 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel. After the completion of electrophoresis, the gel was transferred to the nitrocellulose membrane, and then placed in TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.01% Tween-20) solution containing 5% nonfat skim milk at room temperature for one hour. The membrane was incubated with the primary antibodies diluted to 1:1,000 overnight. Then, the membrane was washed with TBST for 10 minutes three times, and reacted with the secondary antibodies for approximately one hour, followed by washing with TBST for 10 minutes three times. The washed membrane was examined for its band using ECL kit.
Results
1. Effect of the co-administration of mTOR inhibitor and Akt inhibitor
Temsirolimus, a mTOR inhibitor, and GSK690693, an Akt inhibitor, were co-administered to NCI-H460 cells and A549 cells according to indicated concentration. Seventy-two hours after the administration, the cell survival rate was analyzed via MTT analysis. When the cell survival rate was observed by increasing the concentration up to two folds of half maximal inhibitory concentration (IC50), it was shown to have more significantly decreased in the co-administration of temsirolimus and GSK690693 than in the administration of either temsirolimus or GSK690693 alone as the concentration increased (Figure 1A).
To determine objectively whether the aforementioned result was statistically significant, CI was calculated. For NCI-H460 cells, the CI was less than 0.4 at the concentrations of IC50 and 2× IC50, which showed a synergistic effect. Meanwhile, for A549 cells, the CI was less than 0.5 at the concentration of 1/2× IC50 or higher, which also showed a synergistic effect (Figure 1B).
2. Changes in apoptosis after the co-administration of mTOR inhibitor and Akt inhibitor
In the aforementioned synergistic effect of temsirolimus and GSK690693, changes in apoptosis were analyzed with FACS. Forty-eight hours after either co-administration or sole administration of GSK690693 and temsirolimus, FACS was conducted using annexin V staining to analyze apoptosis. For NCI-H460 cells, the apoptosis fraction was shown to be 27.3% in GSK690693 administration, and 11.1% in temsirolimus administration. Meanwhile, it was shown to be 84.5% in the co-administration of GSK690693 and temsirolimus, which showed a significant increase. For A549 cells, the apoptosis fraction was shown to be 14.8% in GSK690693 administration, and 16.6% in temsirolimus administration. Meanwhile, it was shown to be 50.9% in the co-administration of GSK690693 and temsirolimus, which showed a significant increase (Figure 2).
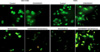
In addition, changes in apoptosis were also examined with TUNEL staining. Forty-eight hours after either co-administration or sole administration of GSK690693 and temsirolimus, staining was conducted and observed using a microscope. As a result, the number of TUNEL-positive cells was shown to have significantly increased in the co-administration of GSK690693 and temsirolimus compared to that observed in the sole administration of either GSK690693 or temsirolimus. The same result was obtained for A549 cells (Figure 3).
3. Changes in autophagy after the co-administration of mTOR inhibitor and Akt inhibitor
PI3K/Akt/mTOR pathway has been known to play an important role in autophagy regulation. Thus, in the aforementioned synergistic effect of temsirolimus and GSK690693, changes in autophagy were also analyzed. When acridine orange staining was conducted to observe the autophagolysosomes, the number of orange color vesicles in the cytoplasm was shown to have significantly increased in the co-administration of GSK690693 and temsirolimus compared to that observed in the sole administration of either GSK690693 or temsirolimus for both NCI-H460 and A549 cells. That is, autophagy was shown to have significantly increased after the co-administration of GSK690693 and temsirolimus (Figure 4).
4. Changes in proteins related to apoptosis and autophagy
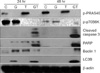
To examine whether the aforementioned synergistic effect of the co-administration of GSK690693 and temsirolimus was also observed in proteins related to apoptosis and autophagy, GSK690693 and temsirolimus were either co-administered or administered alone to NCI-H460 cells, and then western blot was conducted 24 hours and 48 hours after administration. In the examination of the inhibitory effect of GSK690693 on Akt, GSK690693 administration was shown to have inhibited the phosphorylation of PRAS40, a substrate of Akt. In the examination of the inhibitory effect of temsirolimus on mTOR, temsirolimus administration was shown to have inhibited the phosphorylation of p70S6K, a substrate of mTOR. In a relation to apoptosis activation, the cleavage of PARP and caspase 3 was examined after drug administration. Compared to the administration of either GSK690693 or temsirolimus alone, the cleavage of PARP and caspase 3 was shown to have significantly increased after the co-administration of GSK690693 and temsirolimus. In addition, in a relation to autophagy, microtubule-associated protein LC3 type II that reflects autophagy occurrence and beclin 1 expression that is essential for the formation of autophagosome were examined. Compared to the administration of either GSK690693 or temsirolimus alone, LC3 type II was shown to have significantly increased after the co-administration of GSK690693 and temsirolimus. Furthermore, the beclin 1 expression was also shown to have significantly increased 48 hours after co-administration (Figure 5).
5. Effect of the inhibition of apoptosis and autophagy on cell death
To determine the causal relationship between the aforementioned changes in apoptosis and autophagy and cell death, the effect of the inhibition of apoptosis and autophagy on cell death was examined. Fifty µM Z-VAD-FMK, a pancaspase inhibitor, was pretreated as an apoptosis inhibitor, and the drugs were then administered. Forty-eight hours after the administration, the drug-induced cell death was shown to have decreased. The cell survival rate was shown to have changed from 22.64% to 40.78% for A549 cells and from 22.5% to 33.28% for NCI-H460 cells after the co-administration.
In addition, 250µM of 3-methyladenine, an autophagy inhibitor, was pretreated for 4 hours, and the drugs were then administered. Contrary to Z-VAD-FMK pretreatment, the drug-induced cell death was shown to have increased 48 hours after the administration. The cell survival rate was shown to have changed from 22.64% to 7.26% for A549 cells and from 22.5% to 6.78% for NCI-H460 cells after the co-administration (Figure 6).
Discussion
Epidermal growth factor receptor (EGFR) inhibitors have been currently used as useful therapeutic agent for the treatment of lung cancer. However, EGFR inhibitors are effective in only 30% of patients with the activating mutation of EGFR among the total patients. Furthermore, patients who had therapeutic effects show recurrence due to drug resistance. This has been known to be attributable to the second mutation of EGFR causing resistance and the activation of Met signaling pathway which is a bypass of EGFR pathway16. It is interesting to note that as EGFR and Met signal transductions eventually meet in PI3K/Akt/mTOR pathway17, anticancer effect is expected via the inhibition of PI3K/Akt/mTOR pathway despite the second mutation of EGFR related to EGFR resistance or the activation of Met signaling pathway. This is why trials of lung cancer treatment using the inhibition of PI3K/Akt/mTOR pathway draw an attention. Although NCI-H460 and A549 cells used in this study are resistant to EGFR inhibitors, the co-administration of temsirolimus and GSK690693 were shown to be effective in these cells.
In this study, PI3K/Akt/mTOR pathway was simultaneously inhibited by two inhibitors in the lung cancer cells, and the efficacy was then investigated. In previous studies, the efficacy of a single drug was mainly investigated3,4,18. In this study, however, the authors simultaneously inhibited PI3K/Akt/mTOR pathway using two drugs to see whether drug co-administration can increase cell death effect. As a result, the co-administration of temsirolimus, a mTOR inhibitor, and GSK690693, an Akt inhibitor, was shown to have had a synergistic effect through increased apoptosis. The dual inhibition of PI3K/Akt/mTOR is expected to improve the insufficient effect of sole inhibition. In addition, appearance of drug resistance is also expected to decrease in the dual inhibition than in the sole inhibition19. On the other hand, it has been reported that in the sole inhibition of mTOR, negative feedback loops that down-regulate PI3K pathway are also inhibited, which causes the up-regulation of the signaling pathway20. It is also expected to be reduced via dual inhibition. Previous studies, that were conducted on lung cancer cells to investigate the effect of newly developed PI3K/mTOR dual inhibitors, reported that the PI3K/mTOR dual inhibitors improved cell growth inhibition, apoptosis induction, and anti-tumor effect in xenograft model21-24. In this study, cell death caused by apoptosis was shown to have increased by the dual inhibition of PI3K/mTOR pathway, which was consistent with the results of previous studies. Furthermore, autophagy was additionally observed as a mechanism of cell protection in our study.
Autophagy is an essential process for the maintaining of cell survival by decomposing unnecessary proteins or damaged small organelles and using them as energy sources in the cases of stressful conditions such as malnutrition, hypoxia, virus infection, and exposure to toxic materials. As this is, however, a self-limited process, it eventually leads to autophagic cell death if stresses are persistent. Thus, it is called type 2 programmed cell death. That is, autophagy has conflicting both sides in that it can act as a mechanism of both cell survival and death25. In this study, autophagy was shown to have increased after the co-administration of temsirolimus and GSK690693. When the autophagy inhibitor was administered, and the change was then examined, the cell death was shown to have increased. That is, autophagy is likely to be a mechanism of cell protection rather than a mechanism of cancer cell death with respect to the effect of the co-administration of temsirolimus and GSK690693. In the other hand, this result suggests that autophagy could act as a mechanism that is resistant to the effect of Akt/mTOR dual inhibition. In fact, studies are currently under investigation to improve the effect of autophagy inhibitors in a combination of anticancer and radiation therapies in consideration of the cell protection mechanism of autophagy26,27
In this study, a slight difference in the concentrations of temsirolimus and GSK690693 was found between the cells for the synergistic effect. For NCI-H460 cells, the synergistic effect was observed at the concentrations of IC50 and 2× IC50. Meanwhile, for A549 cells, the synergistic effect was observed at a concentration of 1/2× IC50 or higher (Figure 1B). This result is likely to be attributable to difference in drug sensitivity according to cell type. Thus, a further study is required to determine drug concentration in the dual inhibition of PI3K/Akt/mTOR pathway according to cell type.
The results of this study showed that the administration of PI3K/Akt/mTOR inhibitors induced cell death in non-small cell lung cancer, and that the dual inhibition using two drugs improved cell death effect. In addition, autophagy was also shown to have acted as a mechanism of cell protection. A further study is required to explore the possibility of the development of new therapeutic methods based on the results of this study.




 XML Download
XML Download