

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Anti-cancer effects of green tea preparations have been demonstrated in many cancer cell lines and animal models1,2,3, suggesting that green tea may be a cancer preventative. The primary anti-cancer components of green tea are the catechins and epigallocatechin-3-gallate (EGCG) is one of the catechins with the largest biological activity and most frequently demonstrated anti-cancer activity in human and animal models for lung, skin, colon, prostate, and breast cancers3,4,5,6,7.
The biological activities of EGCG include anti-oxidant and anti-inflammatory functions, inhibition of angiogenesis and cancer invasion, and induction of cell cycle arrest and apoptosis8. EGCG can also inhibit telomerase function, which is a target of chemotherapy. However, despite positive results from epidemiological and experimental studies, the effect of EGCG in oncologic practice is not clear. The peak blood level by maximal injection cannot reach inhibitory cancer cell levels in the human body, and a phase I study showed that green tea extract alone was not an effective anti-cancer agent9,10.
Paclitaxel is a member of the taxane family and causes the stabilization of mitotic spindle microtubules by binding the β-tubulin subunit, which leads to G2/M cell cycle arrest and apoptosis11,12. Recent studies of paclitaxel combination treatments have demonstrated that sequential regimens can have a synergistic effect on cancer cell lines13,14,15. We hypothesized that even though EGCG itself may not have any anti-cancer activity, a combination of EGCG with other proven chemotherapeutic agents would have a synergistic effect and that different schedules would differentially affect cancer cells. Therefore, in this study, we investigated the effect of concurrent and sequential combination treatments with EGCG and paclitaxel on growth of NCI-H460, a human non-small cell lung cancer (NSCLC) cell line.
Materials and Methods
1. Cell culture and growth curves
NCI-H460 cells were cultured and maintained in RPMI-1640 media (Sigma Chemical Co., St Louis, MO, USA) supplemented with 10% inactivated fetal bovine serum (FBS) (Sigma Chemical Co.), 100 U/mL penicillin G and 10 µg/mL streptomycin in humidified incubator at 37℃ with 5% CO2. Viable cell growth was analyzed using a 3-[4.5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) assay kit16. The cell seeding number was determined from the exponential growth area derived from cell growth curves.
2. Cell growth inhibition
Harvested H460 cells were plated at a density of 5,000/well in a 96-well tissue culture plate. After overnight incubation, cells were treated with various concentrations of EGCG and paclitaxel in RPMI-1640 with 10% FBS for 72 hours. Following a 72-hour treatment with each agent, cell proliferation assays were performed using a MTT assay kit according to the manufacturer's instructions. The drug concentrations inhibiting cell growth by 50% (IC50) were determined by interpolation from the dose-response curves. Each point represents the mean±standard deviation (SD) of triplicate wells.
3. Combination index assay
The combination index (CI) was calculated to investigate the combined effect of EGCG and paclitaxel. NCI-H460 cells were seeded in 96-well plates at a density of 5,000/well overnight prior to drug treatment. After overnight incubation, the cells were concurrently treated for 72 hours with EGCG and paclitaxel (i.e., E+P), and growth inhibition was measured by MTT assay. For the sequential treatment, after overnight incubation the cells were treated with EGCG first (24 hours), and then were treated with paclitaxel (24 hours) after removal of the media containing EGCG. Finally, they were incubated for an additional 24 hours (i.e., E → P). Growth inhibition was assessed by MTT assay, and the same procedure with the reverse sequence was also performed (i.e., P → E). Each drug combination was tested at four different concentrations, and the CI calculated using a formula reported previously; CI>1 was defined as an antagonistic effect, CI=1 as an additive effect, and CI<1 as a synergistic effect17.
4. Cell cycle analyses
Cell suspensions were plated on 60-mm culture plates and incubated overnight. They were treated with 150 µM EGCG alone, 10 nM paclitaxel alone and their combinations (E+P) for 24 hours. The cells were washed using phosphate-buffered saline (PBS) and fixed with 70% ethanol for 1 hour, and then washed again with PBS and stained with propidium iodide (Molecular Probes, Eugene, OR, USA). Cell cycle analyses were performed using FACScan (Becton Dickinson, San Jose, CA, USA).
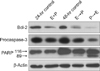
5. Bcl-2, procaspase-3, and PARP expression
Western blotting for Bcl-2, procaspase-3, and poly(ADP-ribose) polymerase (PARP) cleavage were performed to investigate the effect of EGCG and paclitaxel on cell apoptosis. After a 24-hour incubation, cells were treated with 150 µM EGCG and 10 nM paclitaxel concurrently for an additional 24 hours (E+P). In addition, to see the effect of sequential treatments, cells were sequentially treated for 24 hours with each drug (E → P and P → E) after the initial 24-hour incubation.
For protein extraction, cells were washed with PBS and lysed in a RIPA buffer containing protease inhibitors. These lysates were centrifuged at 4℃ for 20 minutes at 14,000 rpm and supernatants containing protein collected. Protein concentrations were determined using the Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA, USA). A total of 30 µg of protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. After transfer to a polyvinylidene fluoride membrane, the blots were blocked with 5% fat free milk in TBS-T (20 mM Tris-HCl, pH 7.4, containing 150 mM NaCl and 0.05% Tween20) and incubated with primary antibodies (1:200, Bcl-2, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; 1:200, procaspase-3, Santa Cruz Biotechnology Inc.; and 1:5,000, PARP, Cell Signaling Technology Inc., Danvers, MA, USA) overnight at 4℃. After washing, blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology Inc.) for 1 hour at room temperature. The protein bands were visualized using an enhanced chemiluminescence kit (GE Healthcare, Little Chalfont, UK).
Results
1. EGCG and paclitaxel inhibit the growth of NSCLC cell lines
The growth of NCI-H460 cells was inhibited by EGCG and paclitaxel in a dose-dependent pattern (Figure 1). The IC50 values of EGCG and paclitaxel were 150 µM and 5 nM, respectively.
2. Combination indices show that sequential paclitaxel → EGCG treatments cause synergistic inhibition of NCI-H460 cell growth
NCI-H460 cells were treated with various concentrations of EGCG (75, 100, 125, and 150 µM) and paclitaxel (1, 2.5, 5, and 7.5 nM) concurrently or sequentially, and growth inhibition curves plotted. The CIs of different concentrations were calculated from the curves (Figure 2). Both concurrent and sequential E → P treatments showed antagonistic effects (CI>1.0) as the dose of each drug increased. However, sequential P → E treatments showed synergistic effects (CI <1.0) when the paclitaxel concentration was >5 nM.
3. EGCG does not have a synergistic effect on cell cycle arrest when concurrently added to paclitaxel treatment
The distribution of cells in different phases of the cell cycle is shown in Figure 3. Treatment with EGCG alone showed a significant but minimal increase in the sub-G1 fraction (p=0.042). In contrast, treatment with paclitaxel alone markedly induced G2/M cell cycle arrest (p=0.001) and increased the sub-G1 fraction (p=0.003). Concurrent (E+P) treatment showed similar results to paclitaxel alone with cell cycle arrest in G2/M. Therefore, EGCG did not seem to have an additive or synergistic effect on cell cycle arrest when added to paclitaxel treatment.
4. Sequential paclitaxel → EGCG treatments improved induction of apoptosis
Western blotting for Bcl-2, procaspase-3, and PARP cleavage was performed with NCI-H460 cells treated with EGCG and paclitaxel (Figure 4). The inhibitory effect was not significant when the cells were treated with both drugs concurrently. However, sequential P → E treatment significantly inhibited the expression of Bcl-2, which was not observed with E → P treatment. Furthermore, Sequential E → P and P → E treatments reduced the expression of procaspase-3, indicating caspase-3 activation, while concurrent treatments showed a borderline effect. In particular, the effect of sequential P → E treatment was significantly greater than that of E → P treatment. Sequential P → E also significantly increased the expression of cleaved PARP activity, compared to other treatment modalities.
Discussion
This study demonstrated that sequential P → E treatment had a synergistic effect on inhibition of NCI-H460 growth when the concentration of paclitaxel was >5 nM. Additionally, EGCG treatment did not have any additive or synergistic effect on cell cycle arrest when concurrently added to paclitaxel. Moreover, western blotting results indicate that sequential P → E treatment can be more effective in the induction of apoptosis, compared to other combination modalities.
There are many studies describing the effect of EGCG on cancer cells. Nihal et al.18 demonstrated that EGCG induced apoptosis in melanoma cells by inhibiting the expression of Bcl-2, increasing the expression of Bax, caspase-3, caspase-7, and caspase-9, and causing S phase and G1 phase arrest. In colorectal cancer cells, Inaba et al.19 showed that, although EGCG increased the phosphorylation of JNK1/2 and ERK1/2 mitogen-activated protein kinases (MAPKs) and increased the expression of caspase-3 and PARP, it inhibited the expression of cyclin D1 and did not affect the cell cycle. This suggested that the effect of EGCG on cancer cells might be associated with MAPK-mediated apoptosis rather than regulation of the cell cycle. Sadava et al.20 investigated the effect of EGCG on small cell lung cancer cells and reported that EGCG induced apoptosis by inhibiting telomerase activity, increasing the expression of caspase-3 and caspase-9, and causing S phase arrest. In NCI-H460 NSCLC cells, our study has demonstrated that EGCG did not have a significant impact on cell cycle arrest, contrary to previous reports. However, the effects of EGCG on the cell cycle can be cell-line dependent and epidemiologic evidence describing the effect of green tea consumption on cancer is conflicting19,20,21,22. Therefore, further studies, including clinical research, are needed to clarify the definite mechanisms and roles of EGCG on cancer cells.
Paclitaxel belongs to the taxane family, which stabilizes microtubules by binding to β-tubulin. The strong inhibitory effect of paclitaxel on lung cancer cells has been well documented; it causes G2/M cell cycle arrest and induces apoptosis by increasing the expression of caspase and decreasing the expression of Bcl-2. Our study confirmed these findings, demonstrating paclitaxel-induced G2/M arrest, decreased expression of Bcl-2 and procaspase-3, and increased PARP cleavage.
While combination treatments are widely used in the treatment of NSCLC, many in vitro studies have determined that their effects vary depending on the sequence in which the drugs are administered. In a study by Shimoyama et al.23, concurrent treatment of gefitinib and camptothecin showed an antagonistic effect on lung cancer cells, while a sequential treatment with camptothecin followed by gefitinib showed a synergistic effect, demonstrated by S phase arrest and increased apoptosis. In a study of paclitaxel, Cheng et al.13 also demonstrated when treating cancer cells sequentially with gefitinib followed by paclitaxel, gefitinib induced G1 phase arrest and as a result, the paclitaxel did not have a sufficient cytotoxic effect. However, when treating cells in the reverse sequence, paclitaxel induced phosphorylation of epidermal growth factor receptor (EGFR) in mitotic cells, which increased the inhibitory effects of subsequent gefitinib treatment. However, several authors suggested that EGCG could also be useful in combination with conventional chemotherapeutics for the prevention and treatment of cancers because it is a non-toxic agent with potential for cancer prevention24. Zhang et al.25 studied the combination effect of EGCG with erlotinib and showed a synergistic effect on squamous cell carcinoma of the head and neck, in which EGCG induced EGFR internalization and ubiquitin-degradation.
In the present study of paclitaxel and ECGC, we found that concurrent treatment (E+P) showed an antagonistic effect (CI>1.0) on growth inhibition but had no effect on apoptosis. This indicates that the effect of concurrent treatment might be inferior to paclitaxel alone and that EGCG could diminish the efficacy of paclitaxel when administered concurrently. However, when EGCG was administered after paclitaxel (i.e., P → E), a synergistic effect was observed based on the CIs and activation of apoptotic cell death was also observed, as demonstrated by decreased expression of Bcl-2 and procaspase-3 and increased PARP cleavage. These results suggest that when adding EGCG to conventional chemotherapy, a sequential treatment model should be considered.
However, in the present study, EGCG inhibited the growth of H460 cells but did not have a significant effect on cell cycle arrest and apoptotic cell death, suggesting that EGCG might inhibit cell growth via different mechanisms. Additionally, we could not demonstrate the exact mechanism that only sequential P → E treatment, not concurrent or sequential E → P models, showed a synergistic effect. Although these can be limitations of the present study, we think that the effects of EGCG can vary depending on the drug concentration and cancer cell line used and that the effect of combination treatments can be schedule-, as well as, time-dependent15. However, we did not vary drug concentration in western blotting nor did we investigate the time-dependent effect in the present study. Therefore, future studies with more detailed methods should be conducted to demonstrate the exact mechanisms.
Our results indicate that the chemotherapeutic effect can be maximized if sequential treatment is used when adding EGCG to other conventional agents. As there are few studies describing the efficacy of EGCG combination treatment with other chemotherapeutics and green tea is a non-toxic anti-cancer agent, the hypothesis tested in this study is relevant. However, before green tea can be used widely in clinical practice, more studies are needed to clarify the molecular interactions between EGCG and other chemotherapeutic agents.



 XML Download
XML Download