

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ataxias are a clinically and genetically heterogeneous group of neurodegenerative disorders. These disorders are accompanied by signs of cerebellar degeneration, but both pyramidal and extrapyramidal features with variable polyneuropathy may also be present.1 Uncoordinated gait, truncal instability, tremors of the body or head, uncontrolled coordination of the hands, dysarthria, and abnormal eye movements are prominent features of cerebellar ataxia.23
Autosomal recessive cerebellar ataxias (ARCA) constitute a highly heterogeneous group of progressive neurodegenerative disorders associated with cerebellar atrophy and spinal tract dysfunction. Friedreich ataxia and ataxia-telangiectasia (A-T) are more common than all other forms of recessively inherited ataxias. A-T is a neuroimmunological disease caused by pathogenic variants in ataxia-telangiectasia mutated (ATM). The ATM gene encodes a serine-threonine kinase that is crucial in DNA repair and is a master regulator in DNA damage responses.4 A-T is a pleiotropic disorder, but its typical features include cerebellar ataxia, telangiectasia, immunodeficiency, hypersensitivity to radiation, and increased risk of malignancies.5 The life spans of A-T patients are usually shortened due to a higher probability of malignancies.6 The absence of full-length ATM is the most common molecular basis of the disorder.78 All reported pathogenic variants of ATM are associated with severe neurological symptoms except for the variant c.5585 T>A (p.Leu1862His); an individual carrying this variant reportedly manifested only mild neurological symptoms.9 In addition, genetic heterozygous carriers of loss-of-function mutations in ATM are at an increased risk of developing breast cancer.10
Ataxia oculomotor apraxia type 2 (AOA2) is a less common form of ARCA that is clinically characterized by progressive cerebellar atrophy, peripheral neuropathy, oculomotor apraxia, and elevated α-fetoprotein levels in serum. The age at onset varies, with people being affected anywhere between 10–20 years of age.11 The gene mutated in AOA2 is SETX. There are at least three isoforms of SETX (which encodes senataxin), of which the longest isoform encodes a protein of 2,706 amino acids. This protein has a C-terminal helicase domain, while the N-terminal domain is important for protein–protein interactions.11 Senataxin is a putative helicase that shares extensive homology with its yeast homologue Sen1. SETX has been suggested to play important roles in the regulation of transcription, mRNA splicing, DNA damage responses, meiotic recombination, normal translational repression, and gene silencing.11121314
AOA2 and A-T are both autosomal recessive disorders associated with dysfunction of DNA repair. Both disorders can be accompanied by oculomotor apraxia, sensorimotor neuropathy, or cerebellar atrophy. AOA2 patients may also have pyramidal signs, whereas A-T patients are at an increased risk of developing cancer. α-Fetoprotein levels are elevated in both disorders, but are significantly higher in A-T patients (150 µg/L in A-T compared to 90 µg/L in AOA2 and less than 10 µg/L in controls). The distinguishing features among the two disorders are telangiectasia, low serum immunoglobin, and younger age at onset in A-T patients (8.2±2.5 years, mean±SD); in AOA2 patients the age at onset is 14.8±2.4 years and the disease progresses more slowly.15
Mutations in SETX have been implicated in two clinically characterized progressive neurodegenerative disorders.11 Dominant gain-of-function mutations in this gene are usually associated with amyotrophic lateral sclerosis (ALS4, OMIM 602433). ALS4 is characterized by atrophy and weakness of distal muscles but without involvement of the sensory system. Two rare phenotypes also reported to be associated with dominant mutations in SETX are tremor ataxia syndrome (TAS)16 and autosomal dominant proximal spinal muscular atrophy (ADSMA).17 TAS patients do not experience peripheral neuropathy but have cerebellar features and tremors, while ADSMA is associated with muscular atrophy, weakness, and increased creatine kinase activity.1118
Recessively inherited mutations in SETX are associated with AOA2 (OMIM 606002), and AOA2 is characterized by cerebellar features along with peripheral neuropathy. Here we report the clinical and genetic characterization of ataxia in two families. A new pathogenic variant in SETX was identified in one of the families, while an ATM pathogenic variant was identified in the second family in two affected individuals and in one asymptomatic child.
METHODS
Ethical compliance and family recruitment
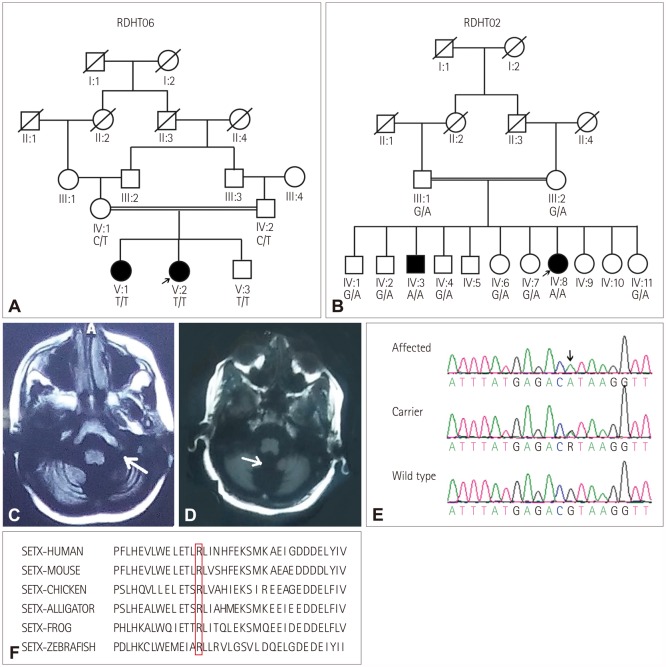
This study was conducted after receiving approval from the Institutional Review Board (IRB No. 00005281) at the School of Biological Sciences, University of the Punjab, Lahore, Pakistan. Families RDHT06 (Fig. 1A) and RDHT02 (Fig. 1B) live in the Punjab province, and two siblings who were offspring of consanguineous parents were affected in each family. The phenotypes of the patients in the two families were distinct, but identical in the pairs of siblings. Written informed consent was obtained from all participants or the parents of young children for inclusion in the study.
Clinical evaluation
Neurologists performed routine and CNS examinations of the patients in both families. The complete blood count (CBC) was measured and magnetic resonance imaging (MRI) of the brain was performed in the patients of both families. Other clinical investigations included electroencephalography (EEG), electromyography (EMG), nerve conduction studies (NCS), electrocardiography, and serum α-fetoprotein levels.
DNA extraction and exome sequencing
DNA was extracted from the blood samples of the participants by a protocol involving sucrose lysis, proteinase digestion, and salting out. A sample from one patient in each family was processed for whole-exome sequencing using the Agilent V4 enrichment kit (Agilent Technologies, Santa Clara, CA, USA), and paired-end reads were obtained at 100× coverage on an Illumina Hi-Seq 2500 sequencer (Macrogen, Seoul, Korea).
Data annotation
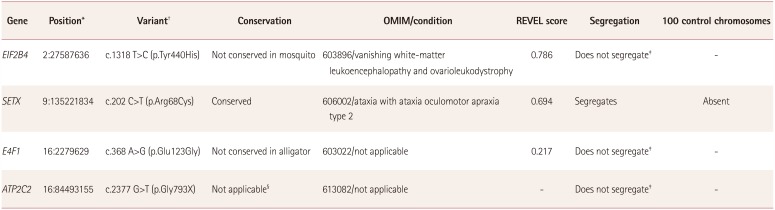
Exome data were annotated using the wANNOVAR application (http://wannovar.wglab.org/). Only homozygous variants were considered given that the mode of inheritance was recessive and the families were consanguineous. Variants were filtered so that those with an allele frequency ≥0.01 in any public database (ExAC, GnomAD, dbSNP, and the 1,000 Genome Project) were excluded. Variants located in the exons and those predicted to affect splicing were considered. The pathogenicity scores predicted by CADD, FATHMM, and PROVEAN were accessed from the wANNOVAR analysis file. In addition, the REVEL score (https://sites.google.com/site/revelgenomics/) was determined for each missense variant. Regions of autozygosity in the whole-exome data were determined using the AgileVCFMapper software (http://dna.leeds.ac.uk/agile/AgileVCFMapper/). Synonymous variants in these autozygous regions were also considered. The conservation of variants in respective orthologues was checked using the HomoloGene site (https://www.ncbi.nlm.nih.gov/homologene), the UCSC multiz alignment tool (http://genome.ucsc.edu/), and by aligning the SETX sequences from different species using the Clustal Omega program (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Segregation analysis and oligos
Specific primers were designed for PCR amplification of regions containing the variants, and the products were subjected to Sanger sequencing using Big Dye Terminators (version 3.1, Thermo Fisher Scientific, Waltham, MA, USA). Segregation analysis was performed on all available family samples, and sequencing results were analyzed with the Lasergene package (DNASTAR Inc., Madison, WI, USA). The frequency of the newly identified pathogenic variant was checked in 100 control chromosomes from an ethnically matched population.
RESULTS
Family 1: ataxia-telangiectasia
The two sisters in family RDHT06 who suffered from ataxia included a 13-year-old female patient (V:2) (Fig. 1A). Her age at the onset of symptoms was 6 years and the disease progressed slowly. Patient V:2 had been bedridden since she was 8-years-old and was unable to sit without support. The patient exhibited choreiform movements, severe dysarthria, and speech difficulties, but did not report any visual or auditory complaints. Her visual acuity was normal and her pupils were of equal size and reactive. Oculocutaneous telangiectasia was present, and the sensory system of the patient was intact. Her intellect was normal and she did not have complaints of urinary or fecal incontinence. Her body tone and bulk were normal. The power was normal (5/5) in the upper limbs,19 but it was only 3/5 in the lower limbs. Deep tendon reflexes were absent. The EEG findings were normal, but MRI showed vermian and cerebellar degeneration (Fig. 1C). Her CBC indicated that she was suffering from anemia. Her older sister had an identical phenotype.
Family 2: ataxia with oculomotor apraxia 2
A 29-year-old female patient (IV:8) in family RDHT02 with two affected individuals (Fig. 1B) suffered from cerebellar ataxia. She was born after an uneventful pregnancy and she achieved normal early milestones. At the age of 20 years her walking became difficult and ataxic, and she complained of progressive weakness in her lower limbs and suffered from frequent falls. She had truncal ataxia, was unable to stand with her feet together, and had a positive Romberg sign. She was unable to perform a tandem gait, instead exhibiting a broad gait with ataxia. She felt a sensation of burning in her feet and also experienced elevations of both heart and breathing rates. She had scanning speech, and eye-focusing difficulties were evident. Her extraocular movements were abnormal and horizontal nystagmus was observed. Slow ocular saccade and diplopia were present. There was no sensory loss, although neuropathy was present. Vertigo and mild-to-severe pulsating headaches were also experienced during walking difficulties with ataxia. Autonomic testing was not performed, but a neurologist found no evidence of autonomic dysfunction. All of the cranial nerves were intact and no facial asymmetry was observed. The power in her limbs was normal, with a negative Babinski sign. However, the tone of the limbs was impaired. No pyramidal signs were observed.
The results of clinical investigations revealed elevated α-fetoprotein, at 51.8 IU/mL (normal range 0.49–9.84 IU/mL). EMG and NCS showed low-amplitude action potentials of motor nerves (data not shown). The hematology report of the patient revealed microcytic anemia. Vermian atrophy was observed in brain MRI (Fig. 1D). No enlarged posterior fossa with torcula-lambdoid inversion was observed. Myelination was normal, and both white matter and gray matter in the brain were intact.
Initially the patient had been misdiagnosed with Dandy-Walker syndrome, even though the triad of radiological features typifying that syndrome were not present. It is possible that the presence of cerebellar vermian atrophy and communication of the fourth ventricle with cisterna magna led to this misdiagnosis (Fig. 1D). General pelvic ultrasonography revealed that her right ovary was somewhat smaller (17 mm) than normal.
The second affected individual (IV:3) (Fig. 1B) had a similar ataxic phenotype as seen in his sister except for having dysarthria and experiencing pain in the eyes during speech. Observation and family interviews revealed that none of the other members of either family had any of the above-described complaints.
Genetic study
The analysis of the filtered exome data (Supplementary Table 1 in the online-only Data Supplement) revealed that the patient in family RDHT06 was homozygous for c.7327 C>T (p.Arg2443Ter, rs121434220), which is a known pathogenic variant in ATM (OMIM 607585, transcript NM_000051.3) and has previously been reported as one of the mutations in an affected individual with compound heterozygous variants.5 Segregation analyses showed that the variant was heterozygous in the obligate carriers of family RDHT06 and also homozygous in the other affected sister. A 5-year-old asymptomatic brother (V:3) of the affected individuals (Fig. 1A) was also homozygous for the variant. A repeated analysis and evaluation confirmed the absence of any manifestation of the disorder in this child. However, since the age at the onset of the symptoms in his siblings was 5–7 years, it is possible that he will develop the disorder later in life.
For the patient in family RDHT02, four variants were identified (Table 1) from the filtered exome data (Supplementary Table 2 in the online-only Data Supplement) that fulfilled the criteria for further analysis. Segregation analysis revealed that variant c.202 C>T (p.Arg68Cys) in SETX (NM_015046.6) was homozygous in the DNA of both affected individuals and segregated with the phenotype classified as spinocerebellar ataxia, autosomal recessive 1 (Fig. 1E). The variant was present at a very low frequency in the ExAC database (0.00001977), with no individual homozygous for the variant. The variant was also absent in 100 control chromosomes from ethnically matched individuals. Four software packages for predicting pathogenicity (SIFT, LRT, MutationTaster, and Polyphen2) predicted this variant to be pathogenic, and the REVEL pathogenicity score for the variant was also high (Table 1). The variant was conserved in the respective orthologues (Fig. 1F). Although the variants c.1318 T>C (p.Tyr440His) in EIF2B4, c.368 A>G (p.Glu123Gly) in E4F1, and c.2377 G>T (p.Gly793X) in ATP2C2 were rare, with frequencies of 0.00003655, 0.002, and 0.001, respectively, and predicted to be pathogenic, they did not segregate with the phenotype (Table 1).
DISCUSSION
DNA damage responses are vital for genomic stability and cell survival.20
SETX and ATM maintain the integrity of the genome by playing roles in oxidative stress and DNA damage repair responses.1121 The variant c.7327 C>T (p.Arg2334Ter) in exon 52 of ATM was previously reported in a compound heterozygous form with a second deletion variant, and both were found to be of African-American origin by comparing ATM mutations in populations with different ethnicities.522 The same variant was also identified as one of the mutant alleles in patients in two different German families. Subsequent haplotype analysis revealed that this region is a mutational hotspot because of a CpG dinucleotide.7 Since a vast majority of individuals in Pakistan have no shared ancestry with either Africans or Germans, observing the same variant in ethnically distinct populations further supports an independent mutational event and confirms that this position is a mutational hotspot.
We identified a novel variant in the protein-interaction domain of SETX as the cause of a progressive disorder of AOA2 in two siblings in a consanguineous Pakistani family. Most SETX variants are recessively inherited and result in AOA2. The characteristic phenotypes of AOA2 include peripheral neuropathy, progressive cerebellar atrophy, oculomotor apraxia, and elevated α-fetoprotein in serum.12 The usual characteristics of cerebellar atrophy, nystagmus, peripheral neuropathy, elevated α-fetoprotein, and weakness in limbs were observed in the present patients. Our patients exhibited radiological findings resembling those seen in Dandy-Walker syndrome, probably due to severe vermian atrophy. The unusual features included mild-to-severe headaches, elevated breathing and heart rates, and the perception of fever in both patients.
SETX as a RNA/DNA helicase is known to play important roles in transcription, neurogenesis, and DNA damage responses.2324 The protein-interaction domain of SETX is located within N-terminal amino acids 64–593, and most pathogenic variants located in this region result in recessively inherited AOA2.25 However, the two variants c.814 C>G (p.His272Asp)26 and c.1166 T>C (p.Leu389Ser)27 in this region are associated with dominantly inherited ALS4 disorder, which makes the genotype–phenotype correlation difficult to establish. Loss-of-function variants that lead to AOA2, and gain-of-function variants that result in ALS4 cause different gene expression profiles in cells, thereby resulting in different phenotypes.28 It is hypothesized that the AOA2-causing SETX variants disturb the sumoylation profile of the gene, whereas ALS4-causing SETX mutations do not. This disruption in sumoylation in turn disrupts the SUMO-dependent interactions between senataxin and the exosome. The disrupted interactions modulate the normal targeting of the exosome to the sites of DNA damage and disturb neuroprotection, which subsequently leads to neurodegeneration.24 A previously identified pathogenic variant in SETX (c.193 G>A, p.Glu65Lys) in the compound heterozygous condition resulting in the AOA2 phenotype25 was reported to disrupt the SUMO-dependent interactions of SETX with the exosome.24 The novel variant c.202 C>T (p.Arg68Cys) identified in the present study is in close proximity to the c.193 G>A (p.Glu65Lys) variant and resides in the protein-interaction domain. It can therefore be hypothesized that this variant can adversely affect the SUMO-dependent interactions of SETX, and verifying this will require further investigations.
This report extends the allelic spectrum of the SETX-associated recessive disorder AOA2. It has also presented the case of a molecularly diagnosed asymptomatic A-T patient for possible recruitment in clinical trials of ATM gene therapy.
 XML Download
XML Download