

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) are classified as thrombotic microangiopathy (TMA), which is characterized by occlusive microvascular thrombosis and by following thrombocytopenia, microangiopathic hemolytic anemia, and organ ischemia.1,2 Acute renal failure is prominent in HUS while neurological impairment and variable degrees of renal abnormalities are found in TTP.1,2 The clinical distinction between HUS and TTP is sometimes unclear because neurologic symptoms can be found in HUS and significant renal insufficiency can develop in TTP.1,2
Deficiency in the von Willebrand factor (VWF)-cleaving protease, also known as ADAMTS13 (a disintegrin and metalloprotease, with thrombospondin 1-like domains motif 13), is causatively related in 70-80% of TTP,3,4 either by compound heterozygous or homozygous mutations in the ADAMTS13 gene in congenital TTP5,6 or circulating inhibitory antibodies in the acquired form.4
However, the focus of research in HUS has been on the mechanisms of injury to the renal endothelium rather than the regulation of VWF.2 In diarrhea-associated HUS (D+HUS), the bacterial agent Shigatoxin from Escherichia coli and Shigella dysenteriae induces the thrombotic state via direct toxic effects, resulting more in renal injury and clinical HUS.7 In diarrhea-negative (D-HUS) or atypical HUS, more than 50% of patients have genetic abnormalities in complement regulatory genes, including complement factor H (CFH), factor I (CFI), membrane cofactor protein (MCP), and factor B (CFB) and C3.8,9 Acquired cases of D-HUS associated with CFH dysfunction due to anti-CFH autoantibodies have also been identified.8
Considering the overlapping clinical manifestation of HUS and TTP, we hypothesized that ADAMTS13 would be an important factor in HUS, and therefore, investigated ADAMTS13 activity as well as ADAMTS13 gene mutation in children with HUS.
The samples of 18 patients with HUS were sent to CHA Bundang Medical Center to test for ADAMTS13 activity and ADAMTS13 gene mutation from April 2004 to August 2005. The diagnosis was based on the presence or absence of diarrhea, renal dysfunction, microangiopathic hemolytic anemia and thrombocytopenia. Eighteen normal controls were recruited from those who visited the Department of Pediatrics of CHA Bundang Medical Center in the same period. In cases of decreased ADAMTS13 activity (less than 44%), the presence of autoantibody in the patient's serum and the ADAMTS13 activity of the parents were tested. The plasma samples were collected from patients and controls with the informed consent, according to the guidelines of the Declaration of Helsinki. The study protocol was approved by the institutional review board of the CHA Bundang Medical Center.
The assay of ADAMTS13 activity was performed as described previously by Furlan, et al.3 Briefly, diluted citrated plasma was activated by barium chloride. This activated plasma was added to protease-free VWF (Green Cross, Yongin, Korea). The reaction was stopped by addition of EDTA. The extent of VWF degradation was assayed by multimer analysis using SDS-electrophoresis in 1.4% agarose gels. Following electrophoresis, the proteins were eletrotransferred to nitrocellulose, and VWF was visualized with horseradish peroxidase-conjugated goat anti-rabbit IgG against human VWF (A0082, Dako, Glostrup, Denmark). The activity was calibrated as previously described10 with 1 : 20 to 1 : 960 dilution of normal human plasma pool.
Inhibition of the ADAMTS13 was assayed by measuring the remaining protease activity in the mixture of patient's plasma and normal pooled plasma at different dilution. Detection of inhibitory activity was carried out by using a screening test, by 1 : 1 mixing pools of normal plasma and patient plasma with reduced ADAMTS13 activity (less than 44%) and by incubating for 30 minutes at 37℃. Thereafter, the mixture was diluted to 1 : 10 in Tris/urea, pH 8.0, and processed further to test ADAMTS13 activity remaining.11
Human genomic DNA was isolated from whole blood. All exons of the ADAMTS13 gene, including the intron-exon boundaries, were PCR-amplified with the primers used by Kokame, et al.6 and Taq DNA polymerase. Products were sequenced in both directions by using a 3700 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Statistics were performed using SAS software (version 8.2) and Chi-square test. p value less than 0.05 was considered statistically significant.
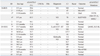
In 18 patients tested, 6 were D-HUS, and 12 D+HUS (Table 1). The median and range for plasma activity of ADAMTS13 in 6 D-HUS and 12 D+HUS patients were 71.8% (22.8-94.1%) and 84.9% (37.9-119.9%), respectively, which were not statistically significantly different from the control group (86.4%, 34.2-112.3%) (p>0.05). No inhibitory activity was detected in patients with ADAMTS13 activity less than 44%.
Five ADAMTS13 gene mutations consisting of 2 novel mutations [1584+2T>A, 3941C>T (S1314L)] and 3 polymorphisms [Q448E, P475S, 2710C>T (S903L)] were found in 3 HUS (2 D-HUS and one D+HUS) patients (Fig. 1). All parents of these 3 HUS patients with ADAMTS13 gene mutations are clinically unaffected. Two novel mutations, 1584 +2T>A, and S1314L, were excluded as common polymorphisms by screening 100 Koreans.
More than 60 mutations of the ADAMTS13 gene have been found in patients with congenital TTP (The Human Gene Mutation Database at the Institute of Medical Genetics in Cadiff, http://www.hgmd.ac.uk/ac/index.php). However, analysis of ADAMTS13 gene mutations has generally been restricted to patients already carrying a clear diagnosis of congenital TTP, potentially presenting an ascertainment bias against the identification of mild mutations.12
While severe deficiency of ADAMTS13 (<10%) establishes a diagnosis of TTP unequivocally,13 not all patients diagnosed with TTP have severe protease deficiency. In addition, ADAMTS13 deficiency alone may not be sufficient to initiate an episode of clinical TTP.14 Patients in clinical remission can still demonstrate ultralarge VWF multimers in the plasma and absence of ADAMTS13 activity in vitro.3 This heterogeneity suggests the presence of genetic modifying factors or environmental triggers other than ADAMTS13 in TTP.1
Complement factor H mutation, known as a causative factor in D-HUS, and Shigatoxin, inducing the thrombotic state in D+HUS, were reported to be risk factors in TTP.9,15 In addition to the overlapping clinical features between TTP and HUS, these findings suggest possible role of ADAMTS13 in HUS.
Decreased ADAMTS13 activity, usually mild to moderate (10 to 40% of normal plasma), has been reported in a wide variety of conditions, including liver cirrhosis, chronic uremia, idiopathic thrombocytopenic purpura, disseminated intravascular coagulation, systemic lupus erythematosus, leukemia, pregnancy, the postoperative state, the neonatal period, and with advancing age16 other than congenital TTP. As for HUS, ADAMTS13 activity is normal or only slightly decreased in typical colitis-associated D+HUS, and severely deficient in a few D-HUS.17
In this study, ADAMTS13 activities were not decreased in 18 HUS patients, which is consistent with previous reports.18 Although the ADAMTS13 activities were normal, we found mutations and polymorphisms of ADAMTS13 gene in 2 D-HUS and one D+HUS patients. We cannot explain the genotype-phenotype dissociation in our 3 HUS patients with ADAMTS13 gene mutations. Heterozygous mutation, distal location, or interaction with polymorphisms might be possible explanations.
One D-HUS (patient 3) patient had heterozygous P475S polymorphism and moderate activity (53.9%) of ADAMTS13 protease. P475S, a well known polymorphism located in the cysteine-rich domain,6,19 was recombinantly analyzed and was found to be associated with a decline in the proteolytic activity of ADAMTS13 (5-10% of wild type) despite normal secretion.20 In Japan, the allele frequency of P475S is about 5%, suggesting that approximately 10% of population are heterozygotes and may possess significantly reduced ADAMTS13 activity.19
The other D-HUS (patient 4) patient had a novel splicing mutation 1584+2T>A in intron 13 (cysteine-rich domain). Also, this patient showed a homozygous S903L mutation in exon 21 (Tsp1-5 domain). Liu, et al.21 reported a highly suspected congenital TTP patient with significantly reduced ADAMTS13 activity and compound heterozygote mutation of S903L and R1095W in the ADAMTS13 gene. Later, S903L was identified as a common polymorphism in Japanese with an allele frequency of 5.5% (7/64).22 Polymorphisms influence the phenotypic expression of complex disease.17 Dependent on the sequence context, the same polymorphisms might be either positive or negative modifiers of gene expression.20 It is unclear whether polymorphism S903L is a positive modifier of ADAMTS13 expression in the context of 1584+2T>A splicing. The interaction between this splicing mutation and S903L polymorphism might explain normal ADAMTS13 activity in this patient.
One D+HUS (patient 7) patient had a novel heterozygous mutation of S1314L on exon 28 CUB2 domain and also heterozygous Q448E. It seems that the location S1314L mutation is too distal to have a detrimental effect on the ADAMTS13 function. However, the cooperative activity between the middle carboxyl-terminal TSP1 repeats and the distal carboxyl-terminal CUB domains is crucial for recognition and cleavage of VWF under flow,23 and more than 10% of ADAMTS13 gene mutations associated with congenital TTP are located in the CUB domains.24 As for Q448E, it is a positive modifier of ADAMTS13 secretion in the context of P618A and A732V, and a negative modifier enhancing the detrimental effect of the missense mutation in the context of R1336W.20 Expression tests would be necessary to elucidate the interaction between S1314L and Q448E.
Limitation of this study is the lack of information on complement system except serum C3 levels. In D-HUS, 4 patients tested C3 levels had normal values. One D+HUS patient had low C3 level with unknown significance.
To our best knowledge, this is the first report about ADAMTS13 gene mutations and polymorphisms in childhood HUS in Korean population. Whether these mutations without reduced ADAMTS13 activity are innocent bystanders or predisposing factors in HUS remains unanswered. In TTP, relapse is common among patients with ADAMTS13 deficiency, but rarely occurs in patients without ADAMTS13 deficiency.25 In 6 D-HUS patients in this study, only the patient 3 and 4 underwent recurrent attack of HUS, suggesting the possibility that ADAMTS13 gene mutations may act as a disease modifying or predisposing factor through an unexplained mechanism.
In the future, expression tests are needed to identify the consequences of each ADAMTS13 gene mutations and polymorphisms found in our HUS patients. Abnormalities in complement system and other genetic or environmental factors, involving TMA, should be investigated. In addition, VWF assay and mutation analysis would be a next assignment in our patients. Long-term follow-up of these HUS patients with ADAMTS13 gene mutations might also provide an invaluable clue.

 XML Download
XML Download