

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In 1977, Carney, et al.1 reported the association between gastrointestinal stromal tumor (epithelioid leiomyosarcoma), functioning extra-adrenal paraganglioma and pulmonary chondroma, and these findings are now referred to as Carney triad. About 100 cases of Carney triad have so far been reported worldwide since its identification. In actual practice, about 78% of patients have just two tumors; most commonly a gastrointestinal stromal tumor and a chondroma.2 Here, we report the first Korean case of Carney triad presenting with a gastrointestinal stromal tumor in the stomach and a malignant functioning paraganglioma in the retroperitoneum.
CASE REPORT
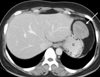
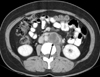
A forty-two years old female patient presented to the hospital because of left lower quadrant abdominal pain and headache. There was no family history of an endocrine tumor or gastrointestinal stromal tumors. Computer tomography showed two separate masses; one was a 5 cm gastric mass with weak enhancement (Fig. 1) and the other a non-enhancing 4 cm-sized left para-aortic mass with lobulated feature (Fig. 2). A needle biopsy of the retroperitoneal mass was performed and the biopsy sample was pathologically diagnosed as a paraganglioma. Since the mass showed multifocal necrosis, a malignant paraganglioma was suggested. The 24 hr urine hormone measurement showed elevated vanillylmandelic acid (VMA), epinephrine, norepinephrin, dopamine, and metanephrine, having 24.3 mg, 202.01 µg, 437.19 µg, 707.29 µg, and 11 mg, respectively. As signs or symptoms of catecholamine excess, hypertension and headache were observed.
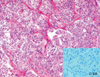
Both tumor masses were removed. The tumor cells in the stomach showed spindle elongated nuclei with pale pinkish cytoplasm (Fig. 3). The tumor showed high cellularity and three mitoses per 50 high power fields, but no necrosis or cellular pleomorphism. The histological features of the retroperitoneal paraganglioma showed a typical Zellballen pattern, and the tumor cells had moderately abundant granular basophilic cytoplasm, which was consistent with a paraganglioma (Fig. 4). These tumor cells were positive for synaptophysin, chromogranin, vimentin, and S-100 protein, but were negative for c-kit and CD34. Metastasis to one of each regional lymph node and cervical lymph node supported the diagnosis of a malignant paraganglioma. Mutation analyses3 performed on both tumors showed no mutations of KIT or PDGFRA. To exclude the possibility of familial paragangliomatosis, further genetic analyses were performed for the SDHB, SDHC and SDHD genes as previously described,4 but these genes were found to be wild. Chest X-ray demonstrated no diagnostic abnormalities.
In spite of surgical removal of the paraganglioma, the 24 hr urine hormones, including VMA, epinephrine, norepinephrine, dopamine, and normetanephrine, remained high and were 16.8 mg, 42.98 µg, 742.95 µg, 426.65 µg, and 6653.38 µg, respectively. Meta-iodobenzylguanidine (MIBG) therapy with 100 mCi was started. Two months after surgery, a hard, tender, non-movable mass was found at the scapular side of right shoulder. Since the mass had grown up to 15 cm and the patient complained of severe pain, the mass was excised. The lesion infiltrated into the subscapular muscle and the infrascapular fossa. The microscopic examination revealed a paraganglioma with diffuse necrosis up to 40% of the tumor volume, and the metastatic tumor involved the bony resection margin. In spite of additional MIBG therapy, a follow-up CT showed omental seeding, liver metastasis, multiple bony metastases, and enlarged retroperitoneal lymph nodes. Combined chemotherapy with cyclophosphamide, vincristine and dacarbazine (CVD) was started. Although the size of the metastatic masses of the omentum, liver and retroperitoneum were decreased after chemotherapy, the patient died of this disease 4 years and 9 months after diagnosis.
DISCUSSION
Carney et al.1 originally reported on the association of gastric epithelioid leiomyosarcoma, pulmonary chondroma, and extra-adrenal paraganglioma in 1977, later referred to as the "Carney triad." Typically, the syndrome affects females (85%) less than 30 years old (82%), who exhibit gastric and pulmonary tumors (75%). Survival for about 20 years after the diagnosis is common, even with hepatic gastrointestinal stromal tumor metastases.2 Carney2 reported that paragangliomas were present in 47% of 79 Carney triad patients. Although the age of onset in our case is included in the whole range of occurrence, clinical onset after 40 years occupies 6% of all the reported cases.2,5-10
Paragangliomas are rare tumors arising from neuralcrest-derived chromaffin cells and may raise the blood pressure by producing adrenalin. As many as 50% of paragangliomas are hereditary and may be associated with familial paraganglioma, neurofibromatosis Type 1, von Hippel-Lindau disease, Carney triad, and rarely with multiple endocrine neoplasia Type 2.11 Although all paragangliomas contain neurosecretory granules, only about 1-3% have clinical evidence of oversecretion. The tumor shows malignant behavior in about 3% of cases and has the ability to metastasize. Familial paragangliomas are often multiple and bilateral, and occur at an earlier age. Mutations in the SDHD (previously known as PGL1), PGL2 and SDHC (previously PGL3) genes have been associated with familial paragangliomas. Patients with Carney triad may develop multiple independent paragangliomas, which may be either "functioning" or "non-functioning." Our case had a functioning malignant paraganglioma which caused headache and hypertension.
In 2002, Carney and Stratakis4 identified a new syndrome which is associated with familial paraganglioma and gastric stromal sarcoma, and they distinguished it from Carney triad. Mutations in SDHB, SDHC and SDHD identified with hereditary paragangliomas were found in these patients.12,13 Although a few familial cases had been reported in the original cohort of patients with Carney triad,2 it was recognized that autosomal dominant inheritance of the dyad of "paraganglioma and gastric stromal sarcoma" or the "Carney-Stratakis syndrome" is a separate condition, which affects both males and females and is not associated with pulmonary chondroma.4 In Carney-Stratakis syndrome, most patients have a family history, gastrointestinal stromal tumor is multifocal, and mutations of SDH genes are found in 73% of cases.12,13 In our patient, the possibility of Carney-Stratakis syndrome was initially considered, however, the absence of a family history, single in numbers of gastrointestinal stromal tumor, and lack of demonstrable mutations in all SDH genes seemed to exclude this possibility and rather suggested a diagnosis of Carney triad.
Although a case with Carney triad was reported to have paragangliomas that were positive for c-kit expression,7 the paragangliomas in our case were negative for c-kit. KIT mutation is usually found in gastrointestinal stromal tumors and rarely in Carney triad,6,8,10 and our case was not an exception.
The prognosis of Carney triad is known to be chronic, persistent and indolent. In a report of 79 cases, only 19% of patients died of disease during follow up, and the mean survival time was reported as 19.6 years.2 However, our case showed very aggressive metastases that were refractory to radioactive iodine therapy and chemotherapy, and the disease led to the death of the patient. The interval between tumor detection and death of the present patient is the shortest ever reported to date. The poor outcome in our case may have been related to the older age of onset or an advanced disease stage at presentation.

 XML Download
XML Download