

 PDF
PDF ePub
ePub Citation
Citation Print
Print


THE HLA SYSTEM
The genetic loci involved in the rejection of foreign organs are known as the major histocompatibility complex (MHC), and highly polymorphic cell surface molecules are encoded by the MHC. The human MHC is called the HLA (Human Leukocyte Antigen) system because these antigens were first identified and characterized using alloantibodies against leukocytes.1 Leukocyte-agglutinating antibodies (leukoagglutinins) were observed in sera from multiparous women and previously transfused patients. Graft rejection was found to be associated with the development of antibodies against allogeneic leukocytes.
The HLA system has been well known as transplantation antigens, but the primary biological role of HLA molecules is in the regulation of immune response.2
Genomic organization of the HLA system
The human MHC maps to the short arm of chromosome 6 (6p21) and spans approximately 3,600 kilobases of DNA.3 The human MHC is divided into three regions (Fig. 1).
The class I region contains the classical HLA-A, HLA-B, and HLA-C genes that encode the heavy chains of class I molecules.
The class II region consists of a series of subregions, each containing A and B genes encoding α and β chains, respectively.4 The DR gene family consists of a single DRA gene and up to nine DRB genes (DRB1 to DRB9). The DRA gene encodes an invariable α chain and it binds various β chains encoded by the DRB genes. HLA-DR antigen specificities (i.e., DR1 to DR18) are determined by the polymorphic DRβ1 chains encoded by DRB1 alleles. HLA haplotypes of certain DRB1 alleles contain specifically linked DRB3, DRB4, or DRB5 locus. The DP and DQ families each have one expressed gene for α and β chains and additional unexpressed pseudogenes. The DQA1 and DQB1 gene products associate to form DQ molecules, and the DPA1 and DPB1 products form DP molecules.
The class III region does not encode HLA molecules, but contains genes for complement components (C2, C4, factor B), 21-hydroxylase, tumor necrosis factors (TNFs), and some others.3
HLA haplotypes
HLA genes are closely linked and the entire MHC is inherited as an HLA haplotype in a Mendelian fashion from each parent. The segregation of HLA haplotypes within a family can be assigned by family HLA studies (Fig. 2). Two siblings have a 25% chance of being genotypically HLA identical, a 50% chance of being HLA haploidentical (sharing one haplotype), and a 25% chance that they share no HLA haplotypes.
Possible random combinations of antigens from different HLA loci on an HLA haplotype are enormous, but certain HLA haplotypes are found more frequently in some populations than expected by chance. This phenomenon is called the linkage disequilibrium. For example, HLA-A1, B8, DR17 is the most common HLA haplotype among Caucasians, with a frequency of 5%.
Expression of HLA
HLA class I molecules are expressed on the surface of almost all nucleated cells. Class II molecules are expressed only on B lymphocytes, antigen-presenting cells (monocytes, macrophages, and dendritic cells), and activated T lymphocytes.
Structure, polymorphism, and function of HLA molecules
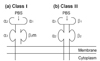
Class I molecules consist of glycosylated heavy chains encoded by the HLA class I genes and noncovalently bound extracellular β2-microglobulin (β2m) (Fig. 3a).5 Human β2m is invariant and its gene was mapped to chromosome 15. The class I heavy chain has three extracellular domains (α1, α2, and α3), a transmembrane region, and an intracytoplasmic domain. The α1 and α2 domains contain variable amino acid sequences, and these domains determine the antigenic specificities of the HLA class I molecules. The α3 and β2m domains together form immunoglobulin constant domain-like folds.6 The heavy chain α1 and α2 domains form a unique structure consisting of a platform of eight antiparallel β strands and two antiparallel α-helices on top of the platform. A groove is formed by the two α-helices and the β-pleated floor, and this is the binding site for processed peptide antigen.2 The class I peptide binding groove accommodates a processed peptide of 8 to 10 (predominantly nonamers) amino acid residues.7,8
The products of the class II genes DR, DP, and DQ, are heterodimers of two noncovalently associated glycosylated polypeptide chains; α and β (Fig. 3b). The α and β chains are transmembrane and they have the same overall structures. An extracellular portion composed of two domains (α1 and α2, or β1 and β2) is anchored on the membrane by a short transmembrane region and a cytoplasmic domain. Polymorphisms of class II molecules occur in the first amino terminal β1 domain of DRB1, DQB1, and DPB1 gene products. The α1 and β1 domains form an antigen-binding groove. The both ends of class II groove are more open that longer peptides (12 amino acids or longer) can be accommodated.9,10
The HLA system is known to be the most polymorphic in humans. The HLA polymorphism is not evenly spread throughout the molecule, but is clustered in the antigen-binding groove.2,5,8 Amino acid variations in several regions change the fine shape of the groove and thus alter the peptide-binding specificity of HLA molecules (see below for details).11 The distribution and frequency of HLA antigens vary greatly among different ethnic groups. It has been postulated that this diversity of HLA polymorphism has evolved under unique selective pressure in different geographic areas. This could be related to the role of the HLA molecules in the presentation of prevalent infectious agents in the different areas of the world.
Zinkernagel and Dougherty demonstrated in 1974 that T lymphocytes must have the same MHC molecules as the antigen-presenting cell to induce immune response.12 The phenomenon that peptides are bound to MHC molecules and these complexes are recognized on the cell surface by the T-cell receptor is called the MHC restriction.
The peptide-binding specificities of HLA molecules are determined by a limited number of amino acid residues located in the peptide-binding pockets.13 Different HLA molecules show characteristic amino acid residue patterns in the bound peptide sequences.11 Conserved amino acid residues located at particular positions of the peptides act as peptide's anchoring residues in the peptide-binding groove.
The nature and source of peptides that will bind to class I or class II molecules are different.10,14 Class I-restricted T cells recognize endogenous antigens synthesized within the target cell (e.g., cellular, transformed, or virus-induced proteins), whereas class II-restricted T cells recognize exogenously derived antigens. The class I molecule-peptide complex on the cell surface is recognized by the T-cell receptor of CD8+ lymphocytes.15 Class II expression is mainly restricted to the antigen-presenting cells including B cells, monocytes/macrophages, dendritic cells, and Langerhans cells. Extracellular exogenous proteins are endocytosed and undergo degradation in the acidic endosomal compartment. The class II molecule-peptide complex is transported to the cell surface, and recognized by the T-cell receptor of CD4+ lymphocytes.16
There are two forms of T-cell receptor (TCR): polypeptide heterodimers composed of disulfide-linked subunits of either αβ or γδ.17 The αβ TCR is present on more than 95% of peripheral blood T cells. During the recognition process between TCR and HLA-peptide complex, accessory molecules on T lymphocytes are enhancing the interaction between T lymphocytes and HLA molecules. The CD4 molecule interacts with a class II molecule on the antigen-presenting cells, and the CD8 molecule interacts with a class I heavy chain on the target cells.
Natural killer (NK) cells are a subset of lymphocytes (10-30% of peripheral blood lymphocytes) that lack TCR and exert cytotoxicity.18 NK cell recognition is not MHC-restricted. NK cells have been known to recognize the loss of expression of HLA class I molecules (i.e., missing self) and destroy cells with decreased expression of class I molecules such as some tumors and virally infected cells. Other cells with normal MHC class I expression can still be NK targets if they provide appropriate signals to activating NK cell receptors. Many different NK cell receptors have been identified and majority of their ligands are HLA class I molecules. NK cells are regulated by both inhibitory and activating signals resulting from the NK cell receptor-ligand binding.18
CLINICAL HLA TESTING
Clinical HLA laboratories perform various tests to support the transplant programs; including HLA typing of the recipient and the potential donor, screening and identification of HLA antibodies in the recipient, and detection of antibodies in the recipient that are reactive with lymphocytes of a prospective donor (i.e., crossmatching).
Serologic typing of HLA antigens
The complement-mediated microlymphocytotoxicity technique has been used as the standard for serologic typing of HLA class I and class II antigens.19 HLA typing sera are mainly obtained from multiparous alloimmunized women, and their HLA specificities are determined against a panel of lymphocytes with known HLA types. Some monoclonal antibody reagents derived from immunized mice are also used.
Peripheral blood lymphocytes (PBLs) express HLA class I antigens and are used for the serologic typing of HLA-A, HLA-B, and HLA-C. HLA class II typing is done with B lymphocytes isolated from PBLs because these cells express class II molecules. HLA typing is performed in multiwell plastic trays with each well containing a serum of known HLA specificity. Lymphocytes are plated in the well and incubated, and complement (rabbit serum as a source) is added to mediate the lysis of antibody-bound lymphocytes.
The nomenclature of the HLA system is formally established by the World Health Organization HLA Nomenclature Committee.4
Molecular typing of HLA alleles
Research has revealed that the extent of HLA polymorphism is far higher than previously known by the number of antigen specificities (Table 1). Serologically indistinguishable variants or subtypes of HLA class I and class II antigens were identified, and these variants are different from the wild type by a very few amino acid substitutions, but these can be functionally distinct and relevant in HLA matching for hematopoietic stem cell transplantation.20 Clinical molecular typing had to be developed in order to differentiate serologically indistinguishable but functionally distinct HLA allelic products.
The polymerase chain reaction (PCR)-based technology is used for clinical HLA typing.21 The first method developed uses sequence-specific oligonucleotide probe (SSOP). For HLA class II typing, the variable exon sequences encoding the first amino terminal domains of the DRB1 and DQB1 genes are amplified from genomic DNA. Based on the HLA sequence database,22 a panel of synthetic oligonucleotide sequences corresponding to variable regions of the gene are designed and used as SSOP in hybridization with the amplified PCR products. As an alternative method, polymorphic DNA sequences can be used as amplification primers, and in this case only alleles containing sequences complementary to these primers will anneal to the primers and amplification will proceed. This second strategy of DNA typing is called the sequence-specific primer (SSP) method. The development of HLA class I allele typing had been much behind that of class II. The class I polymorphism is located in the two domains, α1 and α2 (i.e., requiring amplification of the two exons), and there are many more polymorphic sequences (i.e., requiring more probes or primers) than in the class II polymorphism. These characteristics made it more challenging to develop molecular typing strategies for class I.
Actual DNA sequencing of amplified products of multiple HLA loci is increasingly used as clinical HLA typing in support of the unrelated donor hematopoietic stem cell transplantation.23
HLA alleles are designated by the locus followed by an asterisk (*), a two-digit number corresponding to the antigen specificity, and the assigned allele number. For example, HLA-A*0210 represents the tenth HLA-A2 allele within the serologically defined HLA-A2 antigen family.
HLA antibody screening and lymphocyte crossmatching
Preformed HLA antibodies can be detected by testing the patient's serum against a panel of lymphocytes with known HLA types. The complement-mediated microlymphocytotoxicity technique has been the standard, and the anti-human globulin (AHG) enhancement method provides higher sensitivity.24 This test is called HLA antibody screening and the results are expressed as the percentage of the panel cells that are reactive; this is called the % panel reactive antibody (% PRA). For instance, if 10 of 40 panel cells are reactive with a serum, the PRA is 25%. With a panel of well-selected cells representing various HLA antigens, antibody specificities can sometimes be assigned. This information is particularly important for the prospective organ transplant recipient to predict the chance of finding a compatible or crossmatch-negative deceased donor and to avoid specific mismatched HLA antigens in the donor. When a potential donor becomes available, a final crossmatch is performed between the recipient's serum and the donor's lymphocytes to determine the compatibility. The positive crossmatch results are predictive of the risk of graft rejection.25,26 Antibodies to both HLA class I and class II antigens seem to be detrimental.
Alternative methods based on enzyme-linked immunosorbent assay (ELISA) and fluorescence-based flow cytometry or Luminex® technologies are also available for HLA antibody screening and antibody specificity identification with higher sensitivity and specificity. Lymphocyte crossmatches using flow cytometry offer higher sensitivity and are probably more predictive of allograft rejection risks in certain cases.
THE HLA SYSTEM AND TRANSPLANTATION
HLA-A, HLA-B, and HLA-DR have long been known as major transplantation antigens. Recent clinical data indicate that HLA-C matching also affects the clinical outcomes of hematopoietic stem cell transplantation, but HLA-DQ and HLA-DP do not appear critical.27,28
Both T-cell and B-cell (antibody) immune responses are important in graft rejection.29 T lymphocytes recognize donor cell-derived peptides in association with the HLA molecules on the graft.30 CD4+ T helper cells are activated by antigen-presenting cells (APC) carrying HLA class II molecules. The APCs from either the donor or the recipient can activate the recipient's T cells. The donor's APCs present in the graft cause the "direct" activation of the recipient's T helper cells. The recipient's APCs can acquire alloantigens that are shed from the graft, process into peptides, and present to T helper cells to develop the "indirect" activation. Direct T-cell allorecognition plays an important role in acute rejection and indirect T-cell allorecognition in late onset chronic rejection.
Antibodies bound to the graft fix complement and cause damage to the vascular endothelium, resulting in thrombosis, platelet aggregation, and hemorrhage. Hyperacute rejection occurs in patients who already have antibodies specific to a graft. Natural antibodies against ABO blood group and preformed HLA antibodies induce hyperacute rejection. Natural anti-A and anti-B antibodies cause hyperacute rejection because AB antigens are expressed on endothelial cells of grafts. HLA alloimmunization can be induced by blood transfusions, pregnancies, or transplants. Hyperacute rejection can be avoided in most cases by ABO-identical or ABO-major compatible transplantation and by confirming negative lymphocyte crossmatching.26 Acute rejection is primarily the result of T cell-mediated response. Chronic rejection may be due to antibody and cell-mediated responses.
Solid organ transplantation
Various solid organs can be donated by deceased donors, living related donors, or living unrelated donors. Living donors are mostly for kidneys.
The United Network for Organ Sharing (UNOS) in the United States administers deceased donor organ procurement and allocation, and also monitors national policies for solid organ transplantation.31 Increasing numbers of patients are waiting for deceased donor organs, and the national waiting list on the UNOS as of December 2006 exceeds 94,000.32 The UNOS has developed separate allocation policies for different types of solid organ.33
All potential deceased donor organ transplant candidates are registered with the UNOS. In general, each patient is HLA typed, screened for preformed HLA antibodies, and evaluated for various clinical conditions, and each individual is given a numeric score. The UNOS algorithm for allocating deceased donor kidneys takes into account the HLA matching, time of waiting, HLA alloimmunization status, age, and previous organ donation. Pediatric transplant candidates and those with 80% or higher PRA are given preference. Pretransplant crossmatching is performed by the patient's transplantation program. The presence in the recipient of preformed HLA antibodies reactive with a donor's lymphocytes has been considered a contraindication to kidney transplantation.26 Recently, desensitization protocols have been tried with some success in reducing the levels of preformed donor-specific anti-HLA antibodies and converting a positive crossmatch to negative. These protocols include the administration of intravenous immunoglobulin and use of plasmapheresis.34,35
The benefits of HLA matching are well established in kidney transplantation. There is a clear relationship between the degree of HLA matching and kidney graft survival in transplants from living related donors.36 Better results are obtained from an HLA-identical sibling donor than with HLA-haploidentical parents, siblings, or children. Kidney transplantation from a living unrelated donor shows graft survival superior to deceased donor transplantation (except for six-antigen match) despite a greater degree of HLA mismatch.37 These favorable results are probably due to shorter ischemic time and less renal damage.
The influence of HLA matching on the survival of liver and thoracic organs is yet uncertain, even though there is some evidence that the outcome of heart transplantation may be influenced by the degree of HLA matching. Allocation of livers and hearts is based primarily on medical urgency and waiting time. For heart transplant candidates, initial HLA antibody screening is routine and prospective lymphocyte crossmatching is usually performed for HLA alloimmunized patients. Pretransplant crossmatching is not performed prior to liver transplant because of the urgent need of organs and the uncertain benefits of a crossmatch-negative transplant.
The complex system of solid organ procurement and allocation was developed to increase the supply of organs and ensure their equitable distribution under conditions of limited donor organ supply.33
Allogeneic hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation is used to treat hematologic malignancy, severe aplastic anemia, severe congenital immunodeficiencies, and selected inherited metabolic diseases.38 Source of hematopoietic stem cells had been bone marrow, but mobilized peripheral blood stem cells have become more popular, and the use of umbilical cord blood is expected to grow significantly.39 The main advantage of cord blood transplants is that HLA mismatching is better tolerated with less adverse transplant outcomes. Umbilical cord blood transplantation in adults has been limited by the small number of primitive hematopoietic stem cells in the graft, resulting in delayed engraftment and infectious complications. Initial efforts to expand cord blood progenitors ex vivo have resulted in expansion of mature rather than immature hematopoietic stem cells.
The HLA system is the major histocompatibility barrier in stem cell transplantation, and the degree of HLA matching is predictive of the clinical outcome. HLA mismatch between a recipient and a stem cell donor represents a risk factor for graft rejection/failure and acute graft-versus-host disease (GVHD). The latter is caused by immunocompetent donor T cells contained in the stem cell products. T-cell depletion of donor marrow results in lower incidence of acute GVHD, but higher incidence of graft failure, graft rejection, malignant disease relapse (i.e., loss of the graft-versus-leukemia effect), impaired immune recovery, and later complication of Epstein-Barr virus-associated lymphoproliferative disorders.40,41
The risk of graft rejection or failure is especially higher in patients with severe aplastic anemia because these patients are frequently HLA alloimmunized by multiple blood transfusions prior to transplant and their preconditioning regimen is less intensive or non-myeloablative.42
The best compatible hematopoietic stem cells are from an identical twin and a genotypically HLA-identical sibling. For those who do not have a matched sibling, an alternative related member who is HLA haploidentical and partially mismatched for the nonshared HLA haplotypes may serve as an acceptable donor, but these transplants have higher risks for acute GVHD, graft failure, and mortality.43
When an HLA-matched or partially mismatched acceptable related donor is not available, phenotypically matched unrelated donors can be considered.44 The National Marrow Donor Program (NMDP) was founded in the United States in 1986 to establish a volunteer marrow donor registry and to serve as a source of HLA-matched unrelated marrow donors.45,46 The chance of finding an HLA-matched unrelated donor depends on the patient's HLA phenotype.47 Since there is high diversity of HLA polymorphism among different race groups, there is a different chance of finding a match within different race groups. The NMDP registry now contains more than 6.3 million donors. More than 51,000 cord blood units are also available for transplant through the NMDP. There are international donor registries, and most of these registries share their donors. The NMDP facilitated more than 3,000 unrelated donor stem cell donations in 2006, and the cumulative number of unrelated transplants performed has surpassed 25,000.
Unrelated donor transplants are associated with an increased incidence of acute GVHD and graft failure/rejection compared to HLA-matched sibling transplants. Such an increase results partly from mismatch in HLA alleles and possibly from minor histocompatibility antigens.48-50 For this reason, HLA-A, B, C, and DRB1 allele matching is strongly recommended for unrelated donor transplants.51 Some patients do not find a perfectly allele-matched unrelated donor for multiple loci. A partially mismatched unrelated donor can still be considered for some selected patients. Further studies are needed to better understand the unfavorable effects of mismatches at different HLA loci on graft failure/rejection, GVHD, and survival. NK cell-mediated allorecognition has been associated with graft rejection, acute GVHD, and graft-versus-leukemia reactions. Currently studies are underway to elucidate the potential benefits and risks of mismatches in the NK-cell receptors and their HLA class I ligands, especially HLA-C molecules.52,53
Alloimmunization has to be prevented in all potential stem cell transplant candidates. All transplant candidates and recipients should be transfused with leukocyte-reduced cellular components in order to prevent or reduce the risk of HLA alloimmunization elicited by the donor leukocytes contained in cellular products (e.g., red blood cells, platelets). Transfusion of blood products donated from blood relatives should be avoided for a transplant candidate patient. The minor histocompatibility antigens are inherited independently of the MHC region,54 and thus any transfusions from blood relatives could lead to an exposure to possibly relevant minor histocompatibility antigens.
THE HLA SYSTEM IN TRANSFUSION THERAPY
The HLA system can cause adverse immunologic effects in transfusion therapy. These effects are primarily mediated by HLA antibodies developed against the "passenger" donor leukocytes contained in the cellular blood components. These HLA antibodies induced from previous alloimmunization episodes (pregnancy, transfusion, or transplant) can cause platelet transfusion immune refractoriness, febrile transfusion reaction, and transfusion-related acute lung injury.
HLA alloimmunization
Multiparous women are frequently alloimmunized to HLA and their HLA antibodies may persist or become gradually undetectable. Primary HLA alloimmunization by blood transfusion is caused by the leukocytes normally contained in the cellular blood products.55 HLA antibodies found in alloimmunized patients are frequently broadly reactive. It is more common to find antibody with broad reactivity instead of multiple antibodies of different private specificities in patients with high PRA.
The incidence of HLA alloimmunization following transfusions can vary with the patient's diagnosis and therapy.56 HLA antibodies can be detected in 25% to 30% of transfused leukemic patients and can be present in as high as 80% of aplastic anemia patients. Leukemic patients are usually transfused while receiving intensive chemotherapy, which induces immunosuppression and reduces the incidence of transfusion-induced alloimmunization. Severe aplastic anemia patients who had developed HLA alloimmunization have a higher incidence of graft rejection following stem cell transplantations.57
Leukocyte reduction to less than 5 × 106 in a blood product can prevent or reduce the development of primary HLA alloimmunization.58 Leukoreduction can be achieved for platelet and red blood cell components by the use of third-generation leukocyte reduction filters. Leukoreduced platelet products can also be collected from certain models of apheresis equipment. The wider use of leukocyte-reduced blood products is likely to reduce the number of newly HLA alloimmunized patients from blood transfusions. The incidence of HLA antibody development, however, is not decreased or delayed by the leukocyte reduction in patients with previous pregnancies.59 Most previously pregnant patients appear to develop HLA antibodies by a secondary immune response during transfusion therapy.
Refractoriness to platelet transfusion
Platelet transfusion refractoriness is a consistently insufficient response to platelet transfusions. There are immune and nonimmune causes for poor posttransfusion platelet count increments.60,61 The major nonimmune adverse factors are fever, splenomegaly/hypersplenism, antibiotics (amphotericin B, vancomycin, ciprofloxacin), disseminated intravascular coagulation, infection, sepsis, marrow transplantation, venoocclusive disease, and bleeding at the time of transfusion.
Platelets express platelet-specific antigens and HLA class I antigens. The development of antibodies to these antigens can cause immune destruction of transfused incompatible platelets, resulting in an immune refractoriness to random donor platelet transfusions. When patients are suspected for immune refractoriness, HLA and platelet-specific antibody screening is performed. Definite diagnosis of platelet immune refractoriness is confirmed if antibodies against HLA and/or platelet-specific antigens are detected and non-immune causes of platelet refractoriness are ruled out.
Once the clinical and laboratory diagnosis of immune refractoriness is made, the use of special platelet products is indicated. Most patients who are refractory to random donor platelets because of HLA antibodies respond to HLA-matched platelets.60 Some regional blood centers maintain large pools (several thousands or more) of HLA-A and B typed volunteer apheresis platelet donors. If the specificity of the patient's antibodies can be determined, donors who are negative for corresponding HLA antigens can be selected. Donors who are not perfectly matched with the patients, but homozygous for a given HLA locus can also be used (e.g., patient HLA-A2, 3 and donor HLA-A2 only). HLA-matched siblings or HLA-haploidentical family members can donate platelets by apheresis, but these blood-related donors should not support patient's transfusions prior to a stem cell transplant in order to prevent alloimmunization to minor histocompatibility antigens.
A number of techniques have been tried to determine platelet compatibility.62 Platelet crossmatching using a solid-phase red cell adherence technique has been developed.63 This technique detects antibodies against HLA class I and platelet-specific antigens. Apheresis platelet units are crossmatched with the patient's serum, and crossmatch compatible units are identified. The efficacy of crossmatched platelets may be as good as HLA-matched platelets in some patients.
Since primary HLA alloimmunization caused by platelet transfusions is induced by leukocytes in the product, but not by platelets per se,55 this problem can be prevented or reduced by the use of the third-generation leukoreduction filter. Prevention of HLA alloimmunization is indicated for patients who are expected to need long-term platelet transfusions. Experience of the universal prestorage leukoreduction demonstrated decreased incidence of alloimmune platelet transfusion refractoriness.64
Transfusion-associated graft-versus-host disease
When functionally competent allogeneic T lymphocytes are transfused into an individual who is severely immunosuppressed in cellular immunity, these T lymphocytes are not removed and can mount an immune attack against the recipient's cells, causing transfusion-associated graft-versus-host disease (TA-GVHD). TA-GVHD is not common and typically occurs in patients with congenital or acquired immunodeficiencies or immunosuppression that affects T lymphocytes.
TA-GVHD has also occurred in patients without apparent evidence of immunodeficiency or immunosuppression.65 The majority of these studied cases involved blood products from donors who were homozygous for one or more HLA loci for which the recipient was heterozygous for the same antigen and a different one.66 This relationship can be called a one-way HLA mismatch in the GVHD direction and a one-way HLA match in the rejection direction. As a result, the donor's cells will not be recognized as foreign by the recipient's lymphocytes, while the donor's lymphocytes will recognize the recipient's HLA alloantigens. Other risk factors that appear to predispose to TA-GVHD in immunocompetent patients possibly include fresh blood, donation from blood-related donors, and Japanese heritage.65
Fresh blood contains larger numbers of viable and presumably competent lymphocytes than stored blood. The one-way HLA match more likely occurs when an HLA haplotype is shared by a donor and a recipient (i.e., HLA haploidentical), such as in directed donation from blood relatives and among populations with relatively homogeneous HLA phenotypes.66 The latter possibility may account for the observation that more cases of TA-GVHD have been reported among Japanese patients.
The clinical features of TA-GVHD are similar to those of GVHD following a hematopoietic stem cell transplant; i.e., fever, rash, diarrhea, and liver dysfunction. TA-GVHD is further characterized by prominent pancytopenia due to marrow aplasia. Demonstration of donor-derived lymphocytes in the circulation of a patient with characteristic clinical findings is diagnostic for TA-GVHD. The persistence of donor lymphocytes can be tested by molecular HLA typing, by cytogenetic analysis if a donor and a patient are of different sexes, and by other molecular polymorphisms.
There is no effective treatment for TA-GVHD, and most affected patients die within 3 weeks from complications of infections and hemorrhage.
The primary emphasis in TA-GVHD is prevention.67 Gamma irradiation of cellular blood products is the effective way of inactivating donor lymphocytes. Irradiation is indicated for susceptible patients with various clinical conditions (e.g., congenital immunodeficiencies, hematopoietic stem cell transplants, hematologic malignancies undergoing chemotherapy) and for patients receiving intrauterine transfusion, HLA-matched platelets, or blood components donated from blood relatives.
Febrile nonhemolytic transfusion reaction
Febrile nonhemolytic transfusion reaction (FNHTR) is defined as a temperature rise of more than 1℃ or 2°F during or shortly after the transfusion. Fever can be accompanied by chills, and chills in the absence of fever can be considered as a mild febrile reaction. Fever and chills are the most common transfusion reactions, observed in up to 5% of transfused patients.
FNHTR is caused by either an interaction between the recipient's anti-leukocyte antibodies (usually against HLA antigens and less commonly neutrophil-specific antigens) and donor leukocytes contained in the blood components or pyrogenic cytokines produced in the blood components during storage.
Transfusion-related acute lung injury
Transfusion-related acute lung injury (TRALI) is a rare complication resulting in pulmonary edema.68
TRALI is caused by antibodies against HLA class I and class II or neutrophil-specific antigens.69 Implicated antibodies are usually found in the plasma of transfused blood components.68 Less commonly, antibodies are found in the recipient. The antigen-antibody reaction probably activates complement, resulting in neutrophil aggregation and sequestration in the lungs. The release of neutrophil granules leads to pulmonary vascular damage and extravasation of fluid into the alveoli and interstitium. Demonstration of the donor's antibodies specific against the patient's HLA or granulocyte antigens is direct laboratory evidence for TRALI.
An alternative hypothesis suggests the role of biologically active lipids in the development of TRALI.70
Neonatal alloimmune thrombocytopenia
Neonatal alloimmune thrombocytopenia (NAIT) develops as a result of maternal sensitization to paternally inherited platelet antigens in the fetus. Maternal antiplatelet IgG antibodies cross the placenta and cause fetal and neonatal immune thrombocytopenia. The most commonly implicated platelet-specific antigen is HPA-1a.71 Platelet-specific antigens are generally weak immunogens, and additional genetic factors may influence whether HPA-1a-negative women will develop anti-HPA-1a antibody. Individuals with certain HLA haplotypes with HLA-DRB3*0101 allele are more likely to develop antibodies against HPA-1a antigen.72
Traditionally, it has been thought that only antibodies against platelet-specific antigens cause NAIT. Several case reports, however, suggest that HLA class I antibodies may occasionally be involved.73
HLA AND DISEASE ASSOCIATION
Certain diseases, especially of autoimmune nature, are associated with particular HLA types.74 The association level varies among diseases, and there is generally a lack of a strong concordance between the HLA type and the disease. The exact mechanisms underlying the most HLA-disease association are not well understood, and other genetic and environmental factors may play roles as well.
Among the most prominent associations are narcolepsy with HLA-DQB1*0602/HLA-DRB1*1501, ankylosing spondylitis with HLA-B27, and celiac disease with HLA-DQB1*02. The HLA-A1, B8, DR17 haplotype is frequently associated with autoimmune disorders. Rheumatoid arthritis is associated with a particular sequence of the amino acid positions 66 to 75 in the DRβ1 chain that is common to the major subtypes of HLA-DR4 and DR1. Type I diabetes mellitus is associated with HLA-DR3,4 heterozygotes, and the absence of asparagine at position 57 on the DQβ1 chain appears to render susceptibility to this disease.
Primary or hereditary hemochromatosis (HHC) is one of the most common inherited diseases manifested by an increased absorption of dietary iron, resulting in excess iron deposition in the liver, heart, and endocrine organs and finally organ failure. This disease is determined by an autosomal recessive gene, and up to 10% of the population are heterozygous (carriers) and 0.5% homozygous. Previously the unidentified disease gene had been postulated to be closely linked to the HLA-A locus, especially on the HLA-A3 haplotype. Then nonclassical class I-like HLA-H, also namely HFE, was identified as a hemochromatosis gene.75 HFE is located approximately 5 megabases telomeric to the HLA-A locus. The HFE protein is a transmembrane protein, bound with β2-microglobulin, and expressed in intestinal and liver cells. The α1 and α2 domains interact with the transferrin receptor and regulate iron uptake. The most common mutation responsible for HHC is a single amino acid substitution in the α3 domain of the HFE protein, which causes a loss of the functional protein. Cells become iron-overloaded when there is no HFE to negatively regulate the iron flow into the cell's cytoplasm. HFE mutation analysis has become an important diagnostic tool for HHC.
PARENTAGE HLA TESTING
In parentage testing, genetic markers of a child, biological mother, and alleged father are compared to determine exclusion or nonexclusion of the alleged father.
There are some advantages of using HLA types in parentage testing. The HLA system is inherited in a Mendelian manner and extensively polymorphic; its recombination rate is low; mutation has not been observed in family studies; and antigen frequencies are known for many different ethnic groups. The HLA system, however, does not provide a high exclusion probability when the case involves a paternal HLA haplotype that is common in the particular ethnic group. Molecular techniques using non-HLA genetic systems are widely used,76 and HLA typing is rarely used for parentage testing.
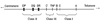


 XML Download
XML Download