

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Caroli's syndrome is a rare congenital disease that consists of intrahepatic bile duct ectasia and congenital hepatic fibrosis. It is believed to be passed down as an autosomal recessive trait. Renal anomalies, such as polycystic kidney diseases, are frequently present as concomitant symptoms. Here, we present a case of an infant who, after being admitted for a urinary tract infection, was diagnosed with Caroli's syndrome and concomitant autosomal recessive polycystic kidney disease (ARPKD).
CASE REPORT
A two month old boy was admitted to our institution with symptoms of a fever, poor oral intake, lethargy, and hazy urine. Initial urinalysis performed in the out-patient department showed many white blood cells in the urine, making the diagnosis of a urinary tract infection a highly likely cause of the boy's ailments. Among blood relatives, there was no known history of genetic, hepatic, or renal diseases. His past medical records were unremarkable. Upon physical examination, the patient was acutely ill-looking and slightly lethargic, but otherwise appeared normal. He suffered from a high fever that exceeded 38℃ and had no detectable hepatosplenomegaly or costovertebral angle tenderness. No icterus was present and his abdomen was unremarkable, without signs of portal hypertensions, such as ascites or distension of abdominal veins. Edema and oliguria were both absent. His weight was 6.3 kg, which was more than the 95 percentile for his age.
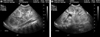
Laboratory studies revealed a hemoglobin level of 9.2 g/dL, leukocyte count of 4420/µL, platelet count of 388,000/µL, total serum bilirubin of 0.4 mg/dL, and direct bilirubin level of 0.2 mg/dL. Serum alanine aminotransferase level was 26 IU/L, aspartate aminotransferase level was 10 IU/L, alkaline phosphatase was 274 U/L, total protein was 4.5 g/dL, and albumin was 3.2 g/dL. Blood urea nitrogen level was 10.9 mg/dL and creatinine level was 0.4 mg/dL. Abdominal ultrasonography (US) was performed for renal evaluation, which revealed a large kidney (Length; Rt - 8.63 cm, Lt - 8.31 cm) with increased cortical echoes and multiple small cysts in the medulla and cortex of both kidneys (Fig. 1). Dilatation of intrahepatic and common bile ducts were noted coincidentally and ARPKD, with Caroli's disease fell under suspicion.
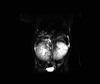
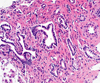
The patient's older brother and both parents were examined ultrasonographically for the presence of renal anomalies, which turned out to be negative. The patient underwent a DMSA scan and a MAG3 diuretic renogram for the functional evaluation of the dysplastic kidneys. The DMSA scan revealed a split renal function of 51.8% on the right and 48.2% on the left kidney. The MAG3 diuretics renogram showed a decrease in both the flow and function of both kidneys. Abdominal MRI showed large, multicystic dysplastic kidneys, fusiform dilatation of the common bile duct, and small, round, and tubular dilatation of the intrahepatic bile ducts (Fig. 2). Needle biopsy of the liver revealed fibrotic changes, a finding that suggested congenital hepatic fibrosis (Fig. 3). Based on these findings, the diagnosis of Caroli's syndrome with ARPKD was made. The patient was put on broad-spectrum antibiotic therapy consisting of ampicillin-sulbactam and cefotaxime. His pyuria persisted, but all his culture studies - including three consecutive urine cultures - proved negative. His fever subsided two days after the initiation of antibiotic therapy and the patient required red blood cell transfusion after the hepatic needle biopsy, but his stay in the hospital was otherwise uneventful. As no overt symptoms remained, the patient was discharged after the fever and other initial symptoms had subsided and is currently being followed up through the out-patient department.
DISCUSSION
Intrahepatic bile ducts develop from bipotential liver progenitor cells in contact with the mesenchyme of the portal vein and thus form the "ductal plates". The ductal plates are then remodeled into mature tubular ducts. Lack of such remodeling results in the persistence of periportal epithelial sleeves, or "ductal plate malformation" (DPM). DPM at different levels of the biliary tree results in variable degrees of fibrosis, while DPM of interlobular bile ducts and associated tubular dilatation of renal collecting tubules results in ARPKD. Congenital hepatic fibrosis may be derived from the same type of liver lesion, via superimposed cholangiopathy that is enough to result in scarring fibrosis. Caroli's disease represents DPM of the larger intrahepatic bile ducts, whereas Caroli's syndrome combines the lesions of Caroli's disease and congenital hepatic fibrosis.1
The term "Caroli's disease" was first described by Jacques Caroli's in 1958 and was later classified as type V choledochal cysts. Mostly, it is inherited as an autosomal recessive trait and can become symptomatic at any age, with males and females being equally affected. Caroli's disease results in communicating cavernous ectasia of intrahepatic bile ducts, with repeated bouts of bile stasis and cholangitis as major symptoms.2 In Caroli's syndrome, the presenting clinical features are similar to Caroli's disease, namely repeated bouts of cholangitis resulting from bile stasis, hepatolithiasis, gallbladder stones, and symptoms associated with hepatic fibrosis, such as portal hypertension and poor hepatic reserve. Cholangiocarcinoma may be present as a late complication in 7% of patients.3,4
Caroli's syndrome is also frequently associated with ARPKD,2,5 which is a rare, inherited disorder that is believed to be caused by a mutation of the PKHD1 gene.6 Its prevalence is approximately one in 6,000 to one in 40,000 newborns,7 and is present as cystic dilatations of the renal collecting ducts. Urine output is usually normal during infancy, but acute oliguric renal failure may ensue with accompanying hypertension. Pulmonary hypoplasia due to oligohydramnios, may also be manifest and if present, can act as a major cause of morbidity and mortality in neonates with ARPKD. Pyuria in the absence of demonstratable bacteriuria, and hyponatremia caused by urine dilution defect are common. Urinary tract infection has also been reported as a common complication.8 When children with ARPKD reach end-stage renal failure, renal transplantation remain as the only curative option. Episodes of urinary tract infections can be managed with support, and hypertensions tend to respond well to angiotensin-converting enzyme inhibitors. Definitive treatment of Caroli's syndrome is also limited, with liver transplantation being the only curative option. In isolated lobar diseases, hepatic lobectomy may also be effective. If concomitant renal failure ensues from the dysplastic kidneys, liver transplantation combined with renal transplantation might be warranted. Acute cholangitis is treated by prompt application of antibiotics and percutaneous transhepatic biliary drainage in adults, with a complication rate of 7-40%,9,10 but its efficacy in infants of our patient's age is not known. Ursodeoxycholic acid is a useful treatment of primary hepatolithiasis in patients with Caroli's syndrome.11
Our patient exhibited ultrasonographic features of ARPKD with pathologically demonstrated hepatic fibrosis, while direct blood relatives were free of renal problems. He did not exhibit any overt symptoms of hepatic or renal dysfunctions at present, enabling us to keep track of his progress and observe him until more radical interventions are warranted. Even though the occurrence of Caroli's syndrome is rare, it should be considered as a possible concomitant disease in asymptomatic patients with polycystic kidney diseases.
 XML Download
XML Download