

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The prevalence of dementia is rapidly increasing in developed countries because of significant increase in aging population. According to the 2014 World Alzheimer's Report, dementia affects approximately 44 million people worldwide, and the incidence of Alzheimer's disease (AD) is expected to triple by the year 2050 (1). Accordingly, the extent of research on the neuroimaging of dementia has expanded tremendously in the last decade, thus enabling easier differential diagnosis of dementia and more precise monitoring of disease progression using structural and physiological magnetic resonance (MR) imaging features. The 3T MRI in particular provides better resolution of structural imaging, and more robust performance of advanced techniques such as diffusion tensor imaging (DTI), perfusion-weighted imaging, MR spectroscopy, and functional MRI. In addition, development of new positron emission tomography (PET) ligands specific for pathological substances such as β-amyloid (Aβ) provides new perspectives on the diagnosis of dementia.
Despite advancements made in MR imaging, radiologists are often reluctant to make a specific diagnosis of clinical dementia due to lack of specific findings. At times, subtle findings such as mild regional atrophy are neglected and imaging diagnosis of dementia is regarded as useless. Therefore, introduction of a systematic and practical approach to the imaging diagnosis of dementia is required. The imaging of dementia has not only diagnostic and prognostic potential, but may also grant unique insights into dementia itself. Therefore, the objective of this review article is to provide a brief overview of AD, and investigate recent neuroimaging developments in the field of dementia with an emphasis on structural MR imaging in order to propose a systematic approach for the imaging diagnosis of dementia.
Alzheimer's Disease
Alzheimer's disease is the most common cause of dementia. AD is characterized by progressive neurodegeneration accompanied by cognitive impairments that interfere with the activities of daily living and ultimately impose heavy economic burdens on the patient and the patient's family (2).
Characteristic Pathology and Pathogenesis Model of AD
Alzheimer's disease is characterized by the accumulation of two abnormal proteins: extracellular Aβ protein and intracellular tau protein (34). Amyloid and tau deposition progress spatiotemporally in a predictive manner. Amyloid first accumulates in the basal part of the frontal, temporal, and occipital lobes, and subsequently spreads to the entorhinal cortex, hippocampus, amygdala, insular cortex, and cingulate cortex, sparing the primary visual and sensorimotor cortices. Conversely, neurofibrillary tangle deposition progresses in the following order: transentorhinal cortex, entorhinal cortex, hippocampus, temporal cortex, association cortices, and finally the primary sensorimotor and visual cortices (5).
The Aβ hypothesis, the dominant theory of AD, suggests that overproduction or inadequate clearance of Aβ is a causative factor for AD; AD begins with the abnormal metabolism of the transmembrane amyloid precursor protein (APP). β- and γ-secretases cleave APPs to form several Aβ peptide fragments (1). Of these, the most important is Aβ42, which is highly prone to aggregation and resultant plaque formation (6). Although amyloid deposits are typically observed in the extracellular space, Aβ is also found within neurons, and this may be related to the aggregation of other cellular proteins such as tau protein in AD (7). Subsequently, abnormal phosphorylation of the microtubule-associated tau protein in neurons and the formation of neurofibrillary tangles are thought to result in the disruption of normal neuronal function (1). Oxidative and inflammatory stresses from Aβ also contribute to the loss of synaptic and neuronal integrity, and finally, neuronal loss and brain atrophy. This downstream pathological cascade has been re-interpreted by the hypothetical model of Jack et al. (8) (Fig. 1). Conversely, Braak and Del Tredici (9) observed hyperphosphorylated tau protein in the absence of Aβ deposition in the medial temporal limbic isocortex of young individuals. Furthermore, recent evidence suggests that tau deposition is a requisite for amyloid toxicity in vivo (10). These findings raise questions regarding the role of Aβ as an initiator of the AD pathophysiological cascade.
Mild Cognitive Impairment or the Prodromal Phase of AD
The time course of AD is approximately 20–30 years from preclinical (cognitive normal) to prodromal (mild cognitive impairment [MCI] or pre-dementia) to overt AD. The prodromal phase of AD is commonly referred to as MCI, which is characterized by the onset of cognitive symptoms (e.g., memory or other cognitive dysfunctions) that do not meet the criteria for dementia (811). The prevalence of MCI in non-dementia individuals over the age of 70 years is approximately 15% with a 2:1 ratio of amnestic to non-amnestic types (1213). Previous studies have indicated that approximately 10–15% of MCI patients progress to AD yearly, and predictors of this conversion include the APOE4 allele of the apolipoprotein E gene (APOE), clinical severity, brain atrophy, certain cerebrospinal fluid (CSF) biomarkers, changes in cerebral glucose metabolism, and Aβ deposition (14). The National Institute on Aging-Alzheimer's Association recently developed new diagnostic criteria for AD, MCI due to AD, and preclinical AD that integrate biomarker evidence into the diagnostic frame (1516). In these criteria, concomitant observation of amyloid deposition (PET or CSF) and neuronal injury (tau, FDG-PET, MRI) indicates a high likelihood of MCI due to AD, while the presence of only one of these factors indicates an intermediate likelihood of MCI due to AD (16). Recent International Working Group-2 criteria further simplified the diagnosis of AD based on the requirement of only the presence of an appropriate clinical phenotype at any stage and presence of amyloid biomarker (e.g., decreased Aβ in CSF, or increased tracer retention on amyloid PET) (17).
Structural Imaging
Structural Imaging as a Mirror of Dementia Pathology
Structural MR imaging is recognized as an important diagnostic tool for AD, as it can both differentiate AD from non-AD dementia and potentially estimate preclinical or prognostic tissue damage in vulnerable regions such as the hippocampus and entorhinal cortex (18). Structural MR imaging is also important for the exclusion of meningioma, glioma, subdural hematoma, vascular malformation, and normal pressure hydrocephalus (19).
Specific patterns of cortical atrophy and vascular pathology distinguish typical AD from other neurodegenerative dementias including frontotemporal dementia (FTD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and vascular dementia (19). Thus, identification and classification of cortical atrophy patterns and vascular pathology are of utmost importance for accurate and effective usage of MR imaging in patients with cognitive impairment.
Cortical Atrophy
Cortical Atrophy in AD and Other Dementias
Accumulation of amyloid plaques and neurofibrillary tangles in AD is contemplated to induce neural and synaptic loss that finally leads to cortical atrophy. Cortical atrophy typically occurs first in the hippocampus and associated entorhinal cortex prior to pervasive progression, and is considered to be an early marker of neurodegeneration (20). Furthermore, the degree of hippocampal atrophy on antemortem MRI has been demonstrated to be correlated with the postmortem severity of neuropathological changes in AD (21). These data suggest importance of accurate recognition of specific atrophy patterns. In the following section, we will discuss specific atrophy patterns and atrophy scales employed for quantitation (Table 1).
Diffuse Cortical Atrophy
Diffuse cortical atrophy is a common finding in various pathological states such as stroke, radiation treatment, and neurodegenerative disorders as well as in normal aging. The global cortical atrophy (GCA) scale was first introduced as a tool to quantify this kind of atrophy in stroke patients with or without dementia. The GCA evaluates atrophy in 13 different brain regions (frontal, parieto-occipital, and temporal sulcal dilation, and dilatation of the ventricles) and assigns a subscore (0 to 3) at each of these levels (22). Studies have demonstrated the predictive value of the GCA for dementia and associated mortality, and confirmed good inter-observer agreement (κ = 0.84) (2324). However, considering that GCA evaluates large brain areas, it is more likely to be confounded by age than other atrophy scales, and is primarily utilized for larger diagnostic assessments (2425). As an alternative, a four-point ventricular enlargement scale (0 to 3) was developed to quantify enlargement of the lateral ventricles in an axial orientation, and this scale has also demonstrated very good inter-observer agreement (intra-class correlation coefficient = 0.85) (26). However, poor diagnostic performance (sensitivity/specificity of 94%/40%) was reported for distinguishing patients who have AD and dementia with Lewy bodies as compared to healthy controls (26). The reason is hypothesized as substantial variation in ventricular size even between healthy individuals (25).
Medial Temporal Lobar Atrophy
Atrophic changes of the medial temporal lobe are commonly found in association with different stages of AD and MCI, but are less commonly related to normal aging (212728). Recently, the NINCDS-ADRDA suggested incorporation of structural MRI of the medial temporal lobe into the criteria for probable AD, but this suggestion did not specify a method for atrophy evaluation.
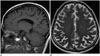
Medial temporal lobe atrophy (MTA) was first quantified by Scheltens et al. (29) using a 5-point subjective scale (0–4) surveying the width of the surrounding CSF spaces and the height of the hippocampal formation; results showed significant differences in MTA between patients with AD and healthy controls in association with good correlation between scale values and linear measurements (Table 2). These findings suggest that visual MTA ratings might provide constructive and rapid assessment for diagnosing or excluding AD in clinical practice (30); however, in a research setting, low inter-observer reliability for this scale (κ = 0.59–0.62) might be problematic (Fig. 2) (3132). Additionally, Scheltens' coronal plane was obtained parallel to the brain stem axis from a midsagittal scout image with a slice thickness of 5 mm and an interslice gap of 1 mm on 0.5 or 0.6T MRI (29), which is different from the current MR acquisition technique to some extent. Recent studies employing the Scheltens 5-point scale with a modified oblique coronal plane perpendicular to the anterior-posterior commissure line with slice thicknesses between 0.8–4 mm have shown more promising results and better inter-observer reliability (weighed κ = 0.65–0.84) (2333).
Duara et al. (34) and Urs et al. (35) also developed a visual rating system for MTA using the mammillary bodies as a landmark to include more standardized portions of the hippocampal head, entorhinal cortex, and perirhinal cortex. Based on this method, standardized atrophy ratings are obtained using software and a 5-point rating scale (0 to 4). This visual rating system showed good diagnostic performance for both MTA and AD (sensitivity/specificity: 82%/82% and 85%/82%, respectively) (3435), but the studies had some limitations. A modified visual rating system for MTA in the axial plane was recently proposed and showed a good correlation with results of the well-known coronal visual rating system designed by Scheltens et al. (κ = 0.772) and fairly good performance for discriminating AD from healthy controls (sensitivity/specificity 76%/80%) (36).
Posterior Cortical Atrophy
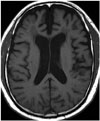
Early posterior cerebral involvement has emerged as an important aspect of AD, and appears to be a feature of early-onset rather than late-onset AD. Therefore, posterior cortical atrophy (PCA) combined with relative sparing of the medial temporal lobe may characterize atypical presentations of AD patients (Fig. 3) (3738394041). The Koedam scale is a 4-point rating scale (0–3) based on the presence of atrophy in sagittal, axial, and coronal orientations of selected anatomical regions: the posterior cingulate sulcus, precuneus, parieto-occipital sulcus, and parietal cortices (42). This scale demonstrated good inter-observer agreement (weighted κ = 0.73) and a sensitivity/specificity of 58%/95% for distinguishing AD; additionally, a subsequent study validated the ability of the Koedam scale to discriminate AD patients from healthy controls (area under the curve [AUC] = 0.74) and frontotemporal lobar dementia patients from AD patients (AUC = 0.66), respectively (38). Recent quantitative validations of this scale have allowed it to be considered as a fast and easily applicable tool in clinical practice (43).
Frontotemporal Lobar Atrophy
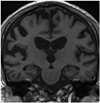
A scale for frontotemporal lobar atrophy (FTLA) was first developed based on the assessment of postmortem specimens of FTD (44), and assessed two coronal slices at the level of the anterior temporal lobe and lateral geniculate nucleus using a 5-point scale (0–4). The scale initially showed potential as an outcome predictor for FTD (Fig. 4) (45). Kipps et al. (46) further extended the scale to include the posterior temporal lobe, and observed a sensitivity of 100% for semantic dementia (SD) and 53% for behavioral variant FTD. It is noteworthy that FTLA scales have been developed with selective focus on the differential diagnosis of FTD, rather than its differentiation from AD.
Anterior Temporal Lobar Atrophy
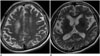
Semantic dementia is a syndromic variant of FTD, and is typified by semantic memory impairment with preservation of episodic memory. While this is a key clinical feature that differentiates SD from AD (47), distinction is not so easily achieved in the earlier stages of disease. One study observed primary involvement of the left anteroinferior and anteromedial temporal lobe with relative sparing of the posterior temporal lobe in SD, as opposed to generalized atrophy in AD (48). Thus, it is proposed that focal anterior temporal lobe atrophy may be a helpful differential diagnostic clue for SD (Fig. 5).
Asymmetric Cortical Atrophy
Bilateral asymmetric perisylvian atrophy can be seen in primary non-fluent aphasia variant of FTD (Fig. 6) (19). CBD produces striking asymmetrical parietal and frontal atrophy while sparing the medial temporal regions, as opposed to near-proportionate atrophy of the fronto-temporo-parietal cortices in AD (Fig. 7) (49). While there are some cases of asymmetrical cerebral atrophy in AD (5051), the observation of asymmetrical cerebral atrophy is usually not in favor of diagnosis of AD.
Quantitative Assessment of Cortical Atrophy
In addition to visual ratings, volumetric and voxel-based measures of brain atrophy have demonstrated close correlations with actual atrophy, neuropathological changes, and cognitive impairment (525354). Given that rates of brain atrophy have been correlated with rates of concurrent cognitive decline, quantitative measurement might serve as an accurate and reproducible surrogate for neurodegenerative pathology (55). However, the labor-intensive nature of such measurements has limited generalization to routine clinical practice (56). Consequently, recent commercial development of automated volumetric measurements of anatomical structures may prove useful in future (5758).
White Matter Hyperintensity
White matter hyperintensity (WMH) has emerged as a strong correlate of cognitive aging and AD (596061). In 1987, Fazekas et al. (62) first described WMH as a periventricular halo, punctate, or early confluent hyperintensity observed in AD. WMH later became known by other terms such as leukoaraiosis, white matter lesion, leukoencephalopathy, and white matter disease, with various definitions that have hampered the integration of previous research on risk factors, pathophysiology, pathological correlations, and clinical implications (63). Recently, the Standards for Reporting Vascular Changes on Neuroimaging (STRIVE) defined WMH as a presumed vascular-origin signal abnormality of variable size in the white matter, showing hyperintensity on T2-weighted images without cavitation, exclusive of subcortical grey matter or brainstem lesions (63).
White matter hyperintensity can be classified as smooth periventricular or irregular periventricular (located in the periventricular system) or deep white matter lesions (found in the subcortical white matter) (6465). Postmortem studies have reported that smooth periventricular WMH presents as a loosening of the fiber network with myelin loss around the tortuous venules, irregular periventricular WMH demonstrates confluent areas with fiber and myelin loss around the perivascular space with reactive gliosis, and deep WMH shows perivascular rarefaction of myelin with varying degrees of axonal loss, astrogliosis, and spongiosis (6667686970). These findings imply that smooth periventricular WMH is likely caused by altered periventricular fluid dynamics, while irregular periventricular WMH and deep WMH reflect ischemic tissue damage (677071). Recent studies have provided strong evidence that WMH plays a causative role in cognitive decline and dementia (72737475), and might lead to AD by contributing in an additive or synergistic manner to disease pathogenesis (597677).
White matter hyperintensity has been considered to be a primary pathology in subcortical ischemic vascular dementia. However, the histopathological features of WMH are also comparable with that of AD, suggesting that subcortical ischemic vascular dementia and AD may be part of a pathological continuum (7879). There are only few pure conditions of AD and subcortical ischemic vascular dementia that have been reported, and rather varying degrees of neurodegenerative and vascular pathologies comprise the spectrum of AD and vascular dementia (80). Furthermore, AD and subcortical ischemic vascular dementia share common risk factors such hypertension, atherosclerosis, diabetes, hypercholesterolemia, obesity, metabolic syndrome, and smoking (80).
The severity of WMH can be quantified semiquantitatively or quantitatively, but a gold standard has not been established until date. The Fazekas scale is most commonly used, which distinguishes periventricular versus deep WMH and scores them according to a 4-point scale (0–3) (Fig. 8) (62). Another well-known scale is the Age-Related White Matter Changes scale, which defines WMHs as ill-defined hyperintensities ≥ 5 mm and scores them according to a 4-point scale in five different regions of each hemisphere (0–30) (81). Fazekas et al. (82) also proposed a WMH grading scale based on type, location, number, and size (0–84). Another new rating system according to the ratio of periventricular versus deep WMHs has been recently proposed by the Clinical Research for Dementia of South Korea (83). These semiquantitative scales have shown good intra-observer and inter-observer agreement and close correlations with volumetric assessments; however, they were designed to measure WMH at a single time point and are not suitable for evaluating WMH progression (848586).
Lacunes
The term "lacune" is derived from the French word for a small fluid-filled cavity, and has historically represented the late stage of a small deep brain infarct (87). The STRIVE criteria define lacunes as a "round or ovoid, subcortical, fluid-filled (similar signal as CSF) cavity measuring between 3 mm and 15 mm in diameter, consistent with a previous acute small deep brain infarct or hemorrhage in the territory of one perforating arteriole" (37). In most cases, lacunes have a CSF-like hypointensity at the center with a surrounding rim of hyperintensity on fluid-attenuated inversion recovery images, which is helpful for differentiating them from enlarged perivascular space along size (< 3 mm) and location criteria (lower basal ganglia region) (888990). Lacunes are assumed to exist as remnants of symptomatic or silent small subcortical infarcts, appearing central to the most common cause of vascular dementia (37). Though there is no consensus regarding the number or location of lacunes required for diagnosis of vascular dementia, two or more lacunes outside the brain stem are considered to be sufficient to support the diagnosis (91).
Lacunes are known to be associated with stroke and gait disturbance in elderly individuals (9293). Furthermore, patients with AD are more likely to have lacunes on MRI than individuals without dementia (7794). Previous pathological studies have also revealed that patients with lacunes are more likely to have dementia for the reason that patients with infarcts require a lower burden of plaques and tangles for clinical diagnosis of AD (959697). Moreover, the location of lacunes is an important factor in dementia, as thalamic and basal ganglia lesions are associated with poor cognition (98). The underlying mechanism relating lacunes and cognitive impairment remains unclear, and may be resultant from the coexistence of other factors such as WMH, cortical microinfarcts, or other unidentified factors (99).
Microbleeds
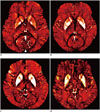
Microbleeds are well-known MRI expression of small vessel disease as well as lacunes and WMH. Cerebral microbleeds refer to small, round, or ovoid hypointensities, ranging 2–10 mm in diameter on T2* gradient-recall echo (GRE) or susceptibility-weighted (SWI) MR sequences, which provide a high contrast-to-noise ratio between the brain parenchyma and paramagnetic materials (100101102). Histopathological-radiological correlation studies have shown that hypointensities found on MRI correlate with areas of hemosiderin deposition with impaired vessel integrity, such as in the case of lipofibrohyalinosis or vascular Aβ deposition in patients with dementia (103104). These hypointensities can be classified into two different patterns of distribution according to their etiology and location: deep or infratentorial location with the presence of vascular risk factors (e.g., hypertension), and lobar (cortico-subcortical) location associated with APOE ɛ4 carrier status or cerebral amyloid angiopathy (CAA) (Fig. 9) (103105106).
With regard to dementia, microbleeds have drawn research attention as potential mediators of cognitive impairment. Microbleeds are more commonly found in patients with MCI, AD, and vascular dementia (107108109110). The mechanism of cognitive impairment due to microbleeds is not fully understood, but is hypothesized to be due to focal dysfunction or damage to the surrounding tissue, or due to more generalized processes such as small vessel disease or extensive CAA (103111). According to the Boston Criteria, for patients older than 55 years who present with lobar intracerebral hemorrhage, the imaging diagnosis of CAA can be made based on either MRI or CT findings: 1) probable CAA: multiple hemorrhage restricted to lobar, cortical, or cortico-subcortical regions; and 2) possible CCA: single lobar, cortical, cortico-subcortical hemorrhage (112).
Microbleeds are also frequently encountered in the context of Aβ immunotherapy for AD (amyloid-related imaging abnormalities) as an important side effect (113). For this particular immunotherapy, Food and Drug Administration guidelines recommend exclusion of patients with more than five microbleeds, which can be evidence of underlying CAA.
Reports of inter-observer agreement on the presence of microbleeds on conventional T2* GRE MR imaging are varied (κ = 0.33–0.88), likely due to detection difficulties (114). Calcification, cavernous malformations, cross-sectional pial vessels, hemorrhagic metastases, and diffusion axonal injury can mimic the appearance of microbleeds on MR imaging, such that a diagnosis should be made in the context of a patient's clinical history and with consideration of other MR sequence findings (63115). Furthermore, T2* GRE versus SWI parameters, resolution, field strength, and post-processing technique can affect the observed number of microbleeds (116117); SWI MR imaging is well-known for detection of a larger number of microbleeds than T2* GRE MR imaging as it offers higher resolution and an increased susceptibility effect (117118119). Therefore, future research should focus on the development and validation of controlled standards for the diagnosis of microbleeds in order to strengthen our understanding of their utility as risk and prognostic factors.
Cerebral Microinfarcts
Emerging evidences shows that cerebral microinfarcts are associated with cardiovascular risk factors as well as cognitive decline and dementia, particularly if they are multiple and cortical (120121). Moreover, cerebral microinfarcts can occur in cognitively normal elderly people, but the prevalence is higher in AD patients (122).
Cortical microinfarcts are defined as sharply defined microscopic regions of cellular death or tissue necrosis, ranging from 50 um to 5 mm as per histopathology studies. Recently, several studies aimed to detect microinfarcts on in vivo 7T MRI (123124). According to a previous study, a cut-off value of five microinfarcts on 7T MRI could differentiate AD and controls with sensitivity of 64.3% and specificity of 94.4% (124). When using 3T MRI, only 25% of cortical microinfarcts identified on 7T were detectable, probably due to limited spatial resolution (123). Consequently, it is necessitated to improve detection of cortical microinfarcts lesion on widely used 3T MRI by protocol optimization to reflect exact burden of microinfarcts in clinical practice.
Systematic Approach to Structural Imaging Evaluation for Differential Diagnosis of Dementia
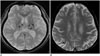
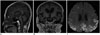
The first thing that must be determined during potential imaging diagnosis of dementia is whether the patient has any focal lesions such as those due to stroke, tumor, or vascular malformation, or if the patient has hydrocephalus (19125). Second, the presence of MTA must be determined, as MTA indicates a high probability of AD. Third, the degree of WMH and the presence of microbleeds should be assessed in order to verify AD or classify the type of dementia (vascular dementia, CAA, and other dementias). If a patient is categorized as having an "other dementia," the spatial pattern of brain atrophy must be more carefully evaluated. FTLA is indicative of FTD. If the FTLA has a right predilection, behavioral variant FTD is indicated, whereas a left predilection indicates SD. Peri-insular atrophy with a left predilection or less commonly, with a right predilection, indicates progressive non-fluent aphasia. Alternatively, presence of parieto-occipital atrophy suggests neurodegenerative diseases such as PCA, CBD, or dementia with Lewy bodies. Infratentorial atrophy with relative sparing pons and prominent atrophied midbrain is indicative of PSP (Fig. 10A) (126). The presence of brain atrophy with markedly disproportionate ventriculomegaly suggests normal pressure hydrocephalus (Fig. 10B) (127). When the cortex and basal ganglia show hyperintensity on diffusion weighted imaging (DWI), Creutzfeldt-Jakob disease is indicated (Fig. 10C) (128). A flowchart of the above approach to the imaging diagnosis of cognitively impaired patients is provided in Figure 11.
Advanced MR Sequences for Detecting Structural Abnormalities
Diffusion Tensor Imaging
In AD, the pathological disruption of cell membranes impedes diffusion of water molecules and produces an increased mean diffusivity, while fractional anisotropy is abnormally decreased in AD and MCI patients due to the loss of tract integrity (129130131). A previous study revealed that DTI measurement can help identify patients with MCI who are likely to progress to AD (132). Moreover, various attempts have been made to assess mean diffusivity and fractional anisotropy from region of interest measurements to tract-based spatial statistics in AD (133134). Axial diffusivity and mean diffusivity are suggested to be most sensitive at detecting early changes of AD (Table 3) (134).
Quantitative Susceptibility Mapping
Quantitative susceptibility mapping (QSM) is an actual quantification map for local susceptibility based on multi-echo 3-dimensional GRE images. QSM can be used to evaluate iron overload potentially associated with neurodegenerative disease (135136). Iron is a known component of Aβ plaques and neurofibrillary tangles, and moreover may provide an ideal environment for Aβ aggregation and neurotoxicity (137138139). Therefore, the use of QSM to measure iron levels in vivo may offer important information to assist our understanding of the neurodegenerative sequence. Indeed, a previous study revealed that significant magnetic susceptibility differences exist in the deep brain nuclei as well as posterior grey and white matter regions of AD patients, and may be a potential biomarker for AD (140). Another study reported the increase of susceptibility (iron load) in vascular dementia as well as AD (Fig. 12) (141).
Magnetization Transfer Imaging
Magnetic transfer (MT) imaging is based on the exchange of magnetization between free protons and protons bound to macromolecules, and reflects underlying histopathological changes in advance than does conventional MRI (142143). Reduced MTR values in the hippocampus have been observed independent of atrophy in even patients with very mild AD (144), thus supporting the contention that MT imaging can detect histopathological changes preceding to obvious volumetric changes in AD. Yet, the underlying mechanism of reduced MT-derived values in the hippocampus of AD patients is not fully understood. One hypothesis is that pathological accumulations of soluble and non-soluble proteins preceding cell death lead to alterations in the local composition of macromolecules, and that the impact of these alterations on MT-derived values is thus more relevant than atrophic changes (145). However, further studies involving larger populations are required to validate the clinical utility and implications of MT imaging in patients with early AD.
Optimized MR Protocols
For the reason that most patients with suspected dementia are elderly and represent a distinct population, a tailored MRI protocol is mandatory. First, imaging markers that reflect important neurohistopathological features of AD should be included. Second, imaging markers that can be used to demonstrate functional changes related to AD should also be incorporated. Third, imaging markers must be identified and utilized to monitor the therapeutic response of AD pathology. Fourth, contrast administration should be avoided unless there are other suspicious imaging findings and MRI should be performed within proper acquisition time. Finally, considering its association with subcortical ischemic vascular dementia, the necessity of MR angiography should be revisited in elderly patients. For MR examinations of patients with suspected dementia, consistency of imaging parameters across subjects and across time are also important in terms of longitudinal follow-up and multicenter clinical trials (146147). We have provided such an MR protocol, which was initially devised for the Korea-Alzheimer Dementia Neuroimaging Initiative (K-ADNI) study by modification of ADNI protocols (Table 4) (148).
CONCLUSION
Imaging of patients with cognitive decline has clear utility not only for exclusion diagnoses but also for the differential diagnosis of neurodegenerative disorders based on specific patterns of atrophy, pathological progression over time, and even physiological changes. Radiologists should be aware of the advantages and limitations of modern imaging so that imaging protocols can be optimized for diagnostic use. Quantitative imaging analysis and diagnosis provides new field and role for radiologists.









 XML Download
XML Download