

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mitochondrial DNA (mtDNA) point mutations have been associated with a wide range of clinical presentations, ranging from pure myopathies to multi-systemic disorders. The G14459A mtDNA mutation changes a moderately conserved alanine residue to a valine within the most evolutionarily conserved region of the ND6 gene, a component of complex I of the mitochondrial electron transport chain (1). The G14459A mutation has been associated with Leber hereditary optic neuropathy (LHON)/pediatric-onset dystonia (2). Eight unrelated families have been identified with this mutation in individuals presenting with LHON, LHON plus dystonia, pediatric-onset dystonia, and clinically asymptomatic phenotype (2-7). In general, LHON presents as an adult-onset acute or subacute visual loss. Pediatric-onset disease associated with the G14459A mutation is characterized by dystonia, short stature, bulbar and corticospinal tract dysfunction, and basal ganglia degeneration on brain magnetic resonance imaging (MRI). The typical age of onset is prior to 5 yr.
Here, we present clinical and molecular data from a Korean family with bilateral striatal necrosis and pediatric-onset dystonia in whom we identified a homoplasmic G14459A mutation in the ND6 gene of the mtDNA. The onset of proband was a 5-yr-old boy and he was severely affected with dysarthria, progressive, generalized dystonia, and spasticity. Brain MRI demonstrated bilateral striatal necrosis. His younger brother, younger sister, nephew, maternal uncle, and maternal cousins had the same clinical features. His mother and elder sister were asymptomatic. None of the family members showed optic neuropathy.
CASE REPORT
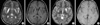
A Korean family was enrolled in this study. The proband (individual III-12), a younger sister (III-13), and a younger brother (III-14) were a 34-yr-old man, a 32-yr-old woman, and a 30-yr-old man, respectively, who had non-consanguineous parents (Fig. 1A). Their health was unremarkable until 5 yr of age, when they developed unilateral distal dystonic posture and gait disturbances. The phenotype progressed and they all currently showed severe dysarthria, contractures of both ankle and wrist, both knee contractures, spasticity in both lower extremity, spinal scoliosis, dystonic hands, and generalized hypotonia, retropulsion, with mild mental retardation. Their clinical features including time of onset were very similar to those of the proband. The proband and younger brother (III-14) was unable to communicate by phone because of severe dysarthria. They could be ambulant by only wheelchair because of multiple joint contractures, spasticity of lower extremity, and dystonia of foot. Vision and hearing were unaffected. Brain MRI showed the presence of bilateral symmetric tissue losses in both the putamen and caudate nucleus (Fig. 2). An increase in lactic acid or amino acids levels in plasma was not observed, and no abnormalities in plasma copper or ceruloplasmin levels were seen. Nerve conduction tests showed no abnormality. The nephew (IV-3), a 6-yr-old boy, exhibited right hand dystonia, right ankle contractures and gait disturbances. However, he was able to walk and swim by himself. He was normally delivered. His onset was at 5 yr old, and it was also similar to that of proband. He was still normal in speech, intelligence, and stature. His MRI revealed bilateral high signal intensity in T2-weighted images, without necrosis (Fig. 2). The maternal uncle (II-3) and maternal cousins (III-1 and III-2) suffered from progressive dystonia. One maternal cousin (III-1) committed suicide in his third decade. The proband's mother (II-7), sister (III-11), and maternal aunt (II-2) were asymptomatic.
Genetic analysis
Six members (II-7, III-11, III-12, III-13, III14, and IV-3) of the family were investigated by molecular genetic tests (Fig. 1A). Four (III-12, III-13, III-14, and IV-3) had progressive, generalized dystonia. Two (II-7, III-11) were asymptomatic. After obtaining informed consent, genomic DNA was isolated from peripheral blood leukocytes using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA), according to the manufacturer's protocol. The samples were analyzed for the presence of 15 point mutations associated with dystonia with bilateral striatal necrosis, LHON, or MELAS (A3243G (7), T3271C, T3308C (8), G3460A, A8296G (9), A8344G, T8356C, G8363A, T8851C (10), T8993G (11), T9176C (11), G11778A, G14459A, T14484C, and T14487C (12) by direct sequencing. In addition, the A3203G mutation, previously identified in a Japanese family, was analyzed (7). The mitochondrial DNA was amplified by polymerase chain reaction (PCR) using primers designed by the authors (available upon request) and a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA). Direct sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems) on an ABI Prism 3100 genetic analyzer (Applied Biosystems). The direct sequencing analysis revealed a homoplasmic G14459A mutation in each of the patients examined (II-7, III-11, III-12, III-13, III-14, and IV-3; Fig. 1B). Thus, two individuals (II-7, III-11) were found to be asymptomatic carriers.
DISCUSSION
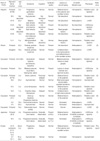
The clinical features of the patients in this Korean family included progressive, generalized dystonia, multiple joint contracture, and spasticity, which were attributable to striatal degeneration. In previous reports, the G14459A mutation was observed in one Hispanic (2), five Caucasian (3-6), one African-American (3), and one Japanese family (7) (Table 1). The general features of previously reported cases were optic neuropathy, progressive generalized dystonia, mild mental retardation, and spasticity. In our family, pediatric-onset dystonia, spasticity, and dysarthria were the major clinical presentations. Based on the clinical features between Caucasian and non-Caucasian in Table 1, racial difference seemed to be not apparent. The most striking difference in our family could be no LHON manifestation such as visual disturbance or optic nerve atrophy. Although delayed manifestation of visual disturbance was reported in Japanese family, which was shown at 38 yr, at least one person of their pedigree had visual disturbance or optic nerve atrophy in previous reports (2-7). Accordingly, we don't know whether any of our family members will have delayed manifestation of optic neuropathy or not.
Among the described cases (26/27) with the heteroplasmic or homoplasmic G14459A mutation in Table 1, age at onset, dystonia, impaired intelligence, and basal ganglia abnormality in neuroimmage was not statistically significant between heteroplasmic and homoplasmic mutation groups (Table 2). The heteroplasmic G14459A mutation has been associated with heterogeneous clinical phenotypes, varying from asymptomatic to dystonia, LHON, or dystonia plus LHON among family members, while the homoplasmic G14459A mutation has primarily been associated with pediatric-onset dystonia, or asymptomatic phenotype (Table 2). Opthalmopathy was more frequently noted in heteroplasmic type (6/9, 66.7%) than homoplasmic type (3/17, 17.6%), and this finding was statistically significant (P<0.012; we regard P<0.05 as statistically significant). Even if, these are limited results because it was done by literature review with inadequate described information, heteroplasmic G14459A is more associated with LHON and homoplasmic G14459A is associated with pediatric-onset dystonia.
Although neither heteroplasmy nor homoplasmy for this mutation is a direct predictor of LHON or a dystonia phenotype, many individuals are essentially homoplasmic for the mutation. Thus, a threshold effect of the mtDNA mutation may contribute to the phenotype of the mitochondrial disease. However, a threshold effect could not explain why some cases with homoplasmic mutation were asymptomatic. Furthermore, the G14459A mtDNA mutation was also found in three unrelated families with Leigh disease (13). This report adds to the clinical heterogeneity associated with the G14459A mtDNA mutation. The heterogeneous clinical features among the family members described here are unlikely to have arisen from different secondary mtDNA mutations among the family members because they were all from the same maternal lineage. Therefore, these findings supports that other nuclear modifier genes are involved in the pathogenesis of the G14459A mutation (5).
In summary, the G14459A mutation is a candidate mutation for maternally inherited dystonia with bilateral striatal necrosis, regardless of optic neuropathy. To our knowledge, this is the first case revealing a mtDNA mutation in a Korean family with a maternally inherited dystonia. Moreover, the clinical manifestations of the homoplasmic G14459A mtDNA mutation, even within the same family, are heterogeneous and support the hypothesis that nuclear genes may play a role in modifying the clinical expression of mitochondrial disease.


 XML Download
XML Download