

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Scleredema, originally described by Buschke in 1902 (1), is a rare sclerodermatosis of unknown etiology, which is characterized by wooden, nonpitting induration of the skin. In general, scleredema first affects the face and neck, and then may spread symmetrically to the shoulders, trunk, arms, and legs: however, the hands and feet are usually unaffected. The disease has occasionally been found in association with a monoclonal gammopathy (MG), in which serum immunoglobulins are usually of the IgG type and the chains are either of the kappa or lambda type (2-6). On the other hand, ankylosing spondylitis (AS) is a chronic systemic inflammatory disorder of the axial skeleton, mainly affecting the sacroiliac joint and spine, and the association of AS with MG has also been described in the literature (7). To the best of our knowledge, there have been no reports on patients with scleredema and AS accompanied with a MG. We here report a male patient with scleredema and advanced AS accompanied with a MG of IgA-kappa protein.
CASE REPORT
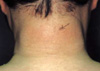
A 40-yr-old man presented with a 7-yr history of a widespread skin thickening, initially noted on the posterior aspect of the neck. He had no history of diabetes mellitus or preceding upper respiratory tract infection. Raynaud phenomenon and dysphagia were not noted. The pain and stiffness in the lower lumbar region and buttock had begun since the age of 24 yr and had been worse in the morning and improved with activity. Physical examination disclosed a symmetric woody induration of the skin on the face, neck, shoulders, trunk, arms, and legs (Fig. 1). His hands and feet were spared. Facial expression was lost: however, the tongue was not enlarged and the frenulum of the tongue was normal. There were no nodules in the skin. In addition, there was marked restriction of lumbar and cervical spine movements. The chest expansion at the fourth intercostal space and modified Schober test were 3.5 cm and 2 cm, respectively. Otherwise, the remaining physical examination was unremarkable.
The blood counts, ESR, calcium, urinalysis, glucose tolerance test, and antistreptolysin O titer were normal. There were no abnormalities in chemistry except that the ratio of albumin vs. globulin was reversed, as albumin 3.7 g/dL vs. globulin 4.4 g/dL. Antinuclear antibody was negative, and HLA-B27 was positive. The IgA level was elevated to 2,084 mg/dL (normal, 70-400): IgG and IgM values were normal. Serum protein electrophoresis revealed a spike in beta globulin fraction, and serum immunoelectrophoresis showed a monoclonal IgA-kappa protein. Bence-Jones proteinuria was absent. Bone marrow examination revealed no evidences for multiple myeloma.
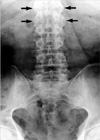
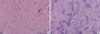
Radiology of the spine and pelvis showed features of advanced AS, including near total ankylosis of bilateral sacroiliac joints and a bamboo spine appearance (Fig. 2). There were no osteolytic lesions on the skeletal survey. Histology of a skin biopsy taken from the arm revealed normal epidermis and a markedly thickened dermis. The collagen bundles were thickened and separated from one another. Alcian blue stain demonstrated abundant mucin deposits between collagen bundles (Fig. 3). The diagnosis of scleredema and AS with a MG was established. After the nature and clinical course of the disease were explained, the patient refused to undergo further treatment, except for nonsteroidal anti-inflammatory drugs.
DISCUSSION
Based on the clinical and histological features, scleredema can be differentiated from other conditions associated with the skin induration, such as systemic sclerosis and scleromyxedema. In contrast to patients with systemic sclerosis, the patients with scleredema usually have no evidences of Raynaud phenomenon, acral sclerosis, and thickening of the frenulum of the tongue, as well as negative ANA test. Although scleromyxedema, another skin condition mimicking scleredema, is also noted to be associated with a MG or multiple myeloma, it progresses acrally and presents characteristic large folds or papules, which are not clinical features associated with scleredema (2, 4, 5, 8, 9). Moreover, the scleredema shows histological features different from other skin diseases, in which the dermis is markedly thickened and collagen bundles are characteristically separated from each other by large fenestration with variable amounts of mucin (5, 10). The clinical and histological findings of our patient were compatible with the scleredema. The diagnosis of AS in this patient was made by the clinical findings and radiological features showing advanced, bilateral sacroiliitis.
Three clinical groups of scleredema have been recognized by Graff (11): in the first group, the disease starts abruptly after an acute upper respiratory tract infection, having a tendency to resolve in a period of months to years; the second group begins insidiously without preceding respiratory tract infection and has longer duration over a period of years; the third group is of a long-standing scleredema associated with severe complicated diabetes mellitus. According to this classification, our patient seems to belong to the second group. In Korea, Lee et al. reported 31 patients with scleredema, in whom most patients showed insidious onset and chronic localized lesions on the nape and upper back, and 16 patients (51.6%) had the accompanied diabetes mellitus (12).
In both scleredema and AS, there have been known associations with MG and less commonly with multiple myeloma (2-7, 13). Hodak et al. documented the following findings in the review of the literature: in patients with scleredema and MG, the scleredema was not preceded by any infectious diseases, nor was it accompanied with diabetes mellitus, and it was usually widespread and long-standing (3). These findings were very similar to the skin conditions of our patient. In the case of scleredema with a MG, serum immunoglobulins are usually of the IgG type, although IgA and IgM have also been reported, and their chains are either of the kappa or lambda type (2, 4-6). Renier et al. reported that seven patients (1.3%) out of 557 patients with AS were found to have MG: 5 patients with IgG, 4 of them of the lambda type, and 2 IgM, both of the kappa type were found; no patients with IgA were observed (7). Interestingly, the current patient had a MG of IgA-kappa protein.
It is unclear whether the coexistence of scleredema and AS is more than coincidental. Mild and moderate elevations of serum IgA levels are frequently noted in AS. Although there is some controversy, high levels of serum IgA may be interpreted as evidence for mucosal infections (14). Moreover, Klebsiella pneumoniae, a normal gut flora, has been suggested to be involved in the pathogenesis of AS (15, 16). Of interest, several cases with AS, who had infectious diseases when the MG was discovered, have been reported (7). However, our patient had no evidences of preceding acute illness, and since the scleredema patients with a MG usually have no preceding infection (3), it is obscure whether the infectious process would contribute to the pathogenesis in the current patient.
To date, the treatment for scleredema remains difficult, although significant improvement was reported recently with the electron-beam therapy (6, 17). As mentioned previously, prognosis of this skin disease is variable: the disease following an acute upper respiratory tract infection has a favorable outcome, whereas the scleredema patients without preceding upper respiratory infection usually have long-standing skin disease (6, 11). Since cases with widespread long-standing scleredema, in whom the finding of MG subsequently progressed to overt multiple myeloma, have been reported (3), a periodic assessment in the current patient will be required.
In summary, we report a patient with both scleredema and advanced AS who had an accompanying MG. The patient has had a widespread, long-standing scleredema developed over 10 yrs after the initial manifestation of AS.
 XML Download
XML Download