

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
KBG syndrome (OMIM #148050) is a rare condition characterized by macrodontia of upper central incisors, distinctive craniofacial features, skeletal anomalies, short stature, and neurological involvement that includes seizure, intellectual disability, and global developmental delay. The diagnosis of KBG syndrome is based primarily on clinical features [1, 2, 3]. Ankyrin repeat domain 11 gene (ANKRD11), encoding ankyrin repeat domain-containing protein 11, is located at chromosome 16q24.3, and it has recently been reported as a causal factor of KBG syndrome [4]. Microdeletion at 16q24.3 encompassing ANKRD11 or heterozygous mutation of ANKRD11 represents the common phenotype of KBG syndrome [4, 5, 6]. The overlapping phenotypical features include variable levels of intellectual disability and facial dysmorphisms. Herein, we report a patient carrying a de novo small deletion in 16q24.3 region corresponding to ANKRD11. We have described the clinical characteristics of the patient in detail and reviewed the clinical and genetic aspects of patients previously reported with microdeletions associated with ANKRD11 or heterozygous mutation of ANKRD11.
CASE REPORT
A 6-yr-old boy was referred to our hospital because of short stature and developmental delay. He was born to non-consanguineous parents at 40 weeks of gestation by normal delivery, and his birth weight was 3,130 g (25th percentile). He was the second child, and his sister had a normal development. He showed developmental delays in speech and motor functions, and he started walking at 20 months of age. He played cooperatively with other children. He had a history of speech therapy for dysarthria at the age of 5 yr. He showed borderline intellectual disability and near-normal social function at 6 yr with a total intelligence quotient of 70 and a social quotient of 97. Psychological assessment revealed mild hyperactivity and attention deficits. However, his behavioral characteristics did not meet the diagnostic criteria for autistic disorder. Physical examination at the age of 6 yr and 6 months indicated that he had a short stature (109.3 cm, less than 3rd percentile). He had macrodontia of permanent dentition and craniofacial dysmorphism, including triangular face, prominent forehead, epicanthal fold, anteverted nostrils, broad mouth, and prominent ears. Brain magnetic resonance imaging showed no abnormalities. Bone X-ray showed narrowing and elongation of both iliac wings and body, small femoral heads, small distal tibial epiphyses and tibiotalar slanting, and delayed bone age. Although electroencephalographic analysis indicated that he had a partial seizure, he did not have a clinical seizure. The clinical features of the patient were consistent with those of KBG syndrome.
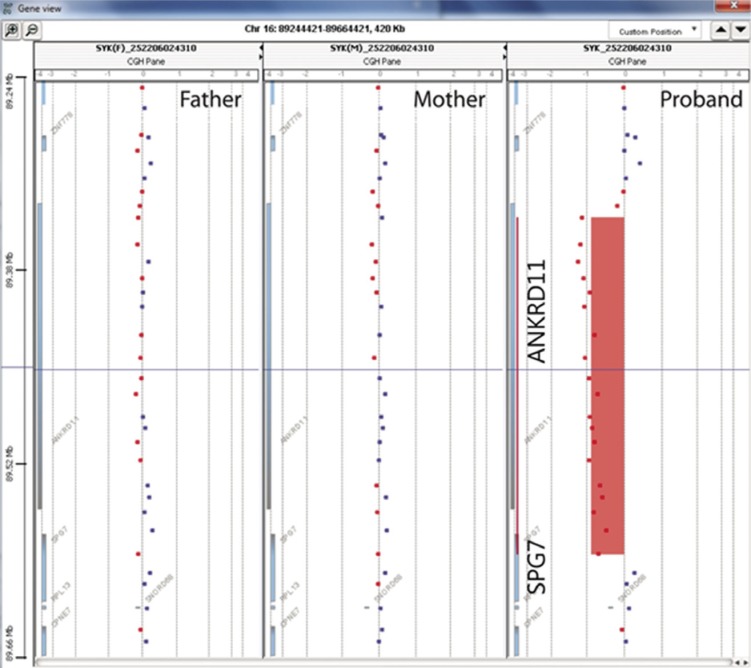
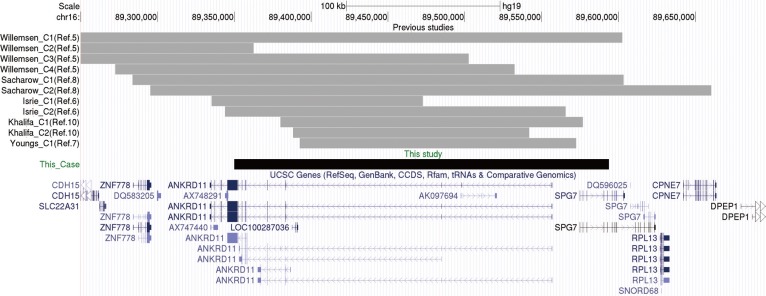
Giemsa/trypsin/Leishman-banded chromosome analysis of the patient and his parents showed an apparently normal karyotype at the 550-band level. Array comparative genomic hybridization (array CGH) was performed by using an Agilent 180K whole-genome oligonucleotide array (version 4.0; Agilent Technologies, Santa Clara, CA, USA), following the manufacturer's protocol. Whole-genome high-resolution oligonucleotide array CGH detected a heterozygous 240-kb deletion at 16q24.3 (arr[hg19] 16q24.3(89,349,966-89,593,853)x1; Fig. 1). Array CGH analysis of parental DNA samples by using the same platform showed no deletion or gain in the 16q24.3 region (Fig. 1). Neither the proband nor the parents had a deletion or gain in any other region. This deletion covered ANKRD11 (including exons 2-13) and part of spastic paraplegia 7 gene (SPG7; including exons 1-5). Fig. 2 shows the genomic map of the deleted region in the proband and in previously reported cases.
This study was approved by the Institutional Review Board of the University of Ulsan College of Medicine and Asan Medical Center, and written informed consent was obtained from the parents of the patient.
DISCUSSION
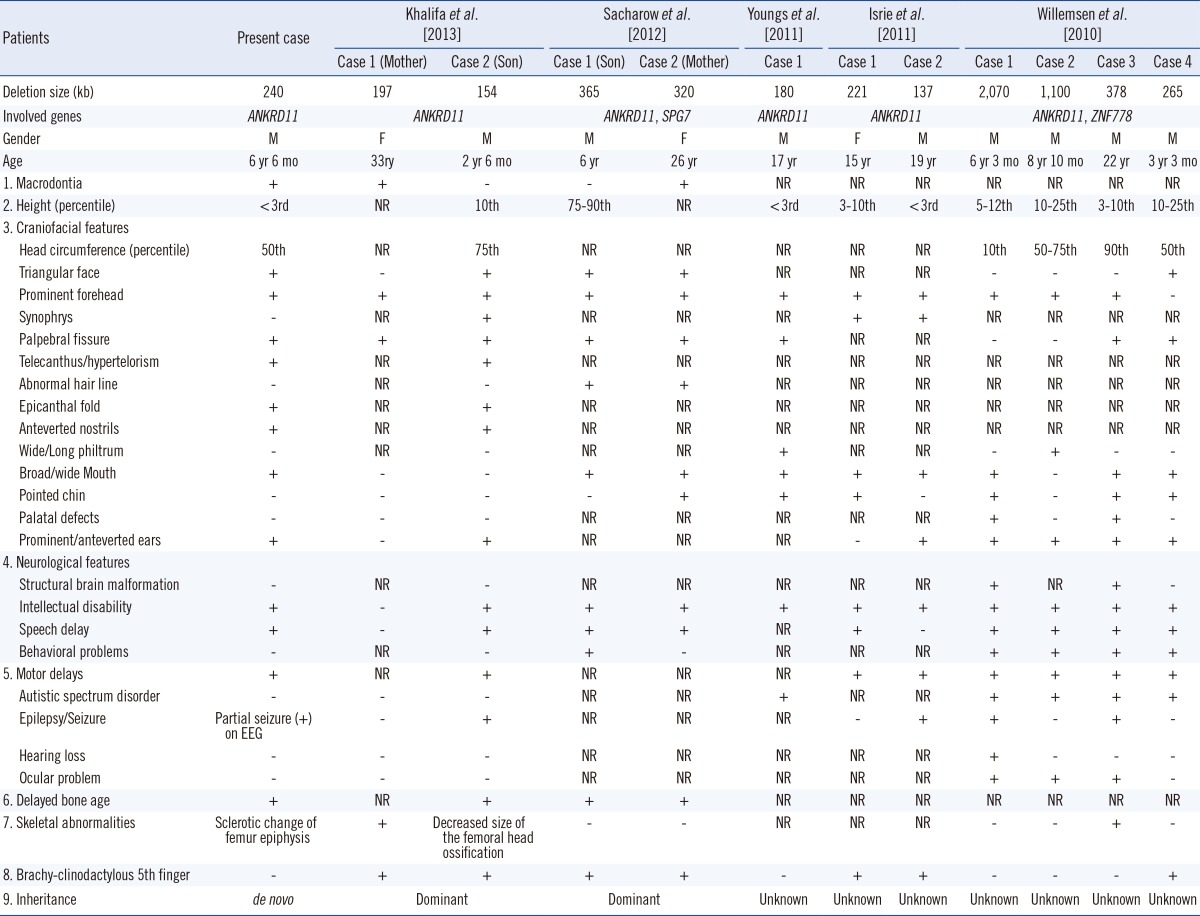
To our knowledge, this is the first report on a Korean patient with KBG syndrome carrying a de novo microdeletion involving ANKRD11. ANKRD11 has been proposed to influence neuronal plasticity by interacting with nuclear receptor complexes and modifying transcriptional activation [4]. Recent studies have reported that patients with heterozygous deletion of ANKRD11 display phenotypes similar to those of KBG syndrome (Table 1) [5, 6, 7, 8, 9]. Our proband displayed deletion of ANKRD11 and partial deletion of SPG7. Mutations associated with SPG7 cause autosomal recessive spastic paraplegia type 7. Hence, the heterozygous partial deletion of SPG7 detected in this patient is probably not related to the features of KBG syndrome.
Willemsen et al. [5] have suggested that this presentation represents the 16q24.3 microdeletion syndrome. Patients with 16q24.3 microdeletion syndrome share the following triad: autism spectrum disorders (ASD), facial and brain anomalies, and variable cognitive impairment. However, in KBG syndrome, intellectual disability, ranging from mild to severe condition, hyperactivity, attention deficit, and easy frustration are more commonly observed than are autistic features [10].
None of the KBG patients with ANKRD11 mutations reported by Sirmaci et al. [4] were diagnosed with ASD. In addition, patient 2 with deletion limited to ANKRD11, described by Isrie et al. [6] showed no autism or ASD. Khalifa et al. [9] have suggested that those patients with deletion in only ANKRD11 lack evidence of autism or ASD. They also suggested that KBG and 16q24.3 microdeletion syndromes are different disease entities. Gene deletion close to 16q24.3 might be involved in autism or ASD. Patients who had mutation or isolated deletion of ANKRD11 showed several common clinical features. Recently, Lo-Castro et al. [11] have reported that intellectual disability was present in all patients with ANKRD11 mutation and ranged from mild to moderate disability. Language development was mostly compromised; however, ASD was absent. Our proband also presented with delayed milestones of moderate degree in language and motor skills, behavioral disturbances such as mild hyperactivity with attention deficit, and no evidence of ASD. These findings suggested that isolated deletion of ANKRD11 region or heterozygous mutation of ANKRD11 is not associated with autism or ASD. In conclusion, our report further suggests that KBG syndrome and 16q24.3 microdeletion syndrome are distinct disease entities.
 XML Download
XML Download