

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Spondyloepiphyseal dysplasia (SED) comprises a heterogeneous group of skeletal dysplasias primarily involving the epiphyses and vertebral bodies. Patients usually exhibit short stature and experience early development of degenerative osteoarthritis. SED has been classified into congenita and tarda forms according to the age of onset and clinical severity. Within the group of SED cases, genetic heterogeneity has been noted and different modes of inheritance in both SED congenita and SED tarda (SEDT) are described [1]. In SEDT, autosomal dominant (MIM 184100), autosomal recessive (MIM 271600), and X-linked recessive (MIM 313400) patterns of inheritance have been reported [2].
The pattern of inheritance and the radiological findings distinguish X-linked (MIM 313400) from autosomal SEDT. Here, we report a 14-yr-old Korean male who presented with a disproportionately short stature and a short trunk, and had a large pedigree of 3 generations with 6 affected family members. Pedigree analysis revealed an X-linked recessive mode of inheritance. In the proband and his mother, a recurrent splice-donor site mutation of the TRAPPC2 (previously called SEDL) gene was identified through direct sequencing analysis. To the best of our knowledge, this is the first report of a genetically confirmed X-linked SEDT case in Korea, and we believe our data highlights the importance of recognizing the mode of inheritance in the diagnosis of X-linked SEDT.
CASE REPORT
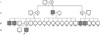
A 14-yr-old Korean male was referred for evaluation of short stature. Upon examination, his height was 147.8 cm (10-25 percentile) and he was 39 kg in weight (10-25 percentile). Short stature was noted in late childhood. He had a disproportionately short stature, a short trunk, and an upper to lower body segment ratio of 0.72. The head circumference was in the normal range and there were no dysmorphic facial features. Motor and cognitive functions were normal. The patient did not complain of hip pain or back pain and there was no evidence of kyphosis and scoliosis. According to the family history, both parents and one brother were unaffected. Five males on the maternal side, however, had short stature persisting over 3 generations, and there was no male-to-male transmission of the phenotype. Therefore, the pedigree analysis strongly suggested an X-linked recessive mode of inheritance of the disease (Fig. 1).
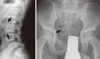
X-ray radiography of the lumbar vertebrae and epiphyses revealed the typical characteristic features of X-linked SEDT (Fig. 2). Platyspondyly, with hump-shaped central portions of the vertebral bodies was noted, and the epiphyses were irregular with flattening of the femoral heads. The clinical diagnosis of X-linked SEDT was based on these radiological features and the 3-generation family history that was indicative of an X-linked recessive mode of inheritance.
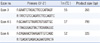
Molecular genetic testing of the TRAPPC2 gene, the only gene associated with X-linked SEDT, was performed on the proband and his unaffected mother. After obtaining their consent, blood samples were collected for DNA extraction and mutation analysis. Genomic DNA was isolated from the peripheral blood leukocytes using the QIAmp DNA Mini Kit (Qiagen, Hamburg, Germany). The DNA was quantified spectrophotometrically using a ND-1000 (Nanodrop Technologies Inc., Wilmington, DE, USA). All 4 coding exons of TRAPPC2 with their immediate flanking intronic regions were amplified using previously reported primer sequences with modifications [3] (Table 1). For all amplicons, the genomic DNA was denatured at 96℃ for 5 min, followed by 35 cycles of denaturation at 96℃ for 30 sec, annealing at different temperatures according to the melting temperature (Tm) of each primer set (Table 1), and extension at 72℃ for 1 min, and a final extension at 72℃ for 5 min. The PCR products were examined by agarose gel (1.5%) electrophoresis followed by ethidium bromide staining (0.5 µg/mL) and were visualized under ultraviolet (UV) light in a gel documentation system (Gel Doc 1000; Biorad, Hercules, CA, USA). PCR amplicons were bidirectionally sequenced with the Big Dye terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) using the ABI 2012.32.3.234PRISM 3100 Genetic Analyzer (Applied Biosystems). RefSeq ID: NM_ 001011658.2 was used for alignment.
Direct sequencing of the PCR products revealed hemizygosity for a splice-donor site mutation in intron 3 of the TRAPPC2 gene (c.93+5G>A) in the proband and heterozygosity for the same mutation in his unaffected mother (Fig. 3).
DISCUSSION
X-linked SEDT is a rare disease with an estimated prevalence of 1.7 per 1,000,000 [4]. A common presenting feature of X-linked SEDT is a disproportionate (short trunk) short stature due to platyspondyly, commonly described as being "barrel-chested". Also, dysplasia of the weight-bearing joints often renders replacement of the hip joints necessary in the third decade of life. At birth, the affected males are normal in length and normal in body proportions, however they exhibit delayed linear growth beginning around 6-8 yr of age, and the usual age of presentation is after the first decade of life.
In this case, an X-linked recessive inheritance pattern was strongly suspected based on pedigree analysis and the absence of male-to-male transmission of the phenotype. The radiological manifestations most characteristic of X-linked SEDT include platyspondyly with anterior wedging, a central and posterior hump of the vertebral bodies, and narrow and irregular inter-vertebral spaces. In the adult, the vertebral changes are diagnostic, but in early adolescence, radiographic findings may be confused with other diseases including mild Morquio's disease, multiple epiphyseal dysplasias with vertebral changes, and adolescent kyphosis [5].
The TRAPPC2 gene that is responsible for X-linked SEDT was identified by Gedeon et al. in 1999 [3]. TRAPPC2 is a widely expressed and highly conserved gene localized to Xp22 [6], consists of 6 exons, and spans a genomic region of approximately 20 kb. The coding regions are located on exons 3-6, and the gene encodes a 140-amino acid protein (sedlin) that has a putative role in endoplasmic reticulum (ER)-to-Golgi vesicular transport [3]. Forty-seven different TRAPPC2 mutations across the open reading frame (ORF) have been reported so far [7], which are spread over the entire coding region of the TRAPPC2 gene in exons 3-6; however, approximately 90% of these mutations involve exons 4, 5, and 6 [1]. The most common type of disruption in TRAPPC2 gene is deletion, accounting for nearly 50% (23/47) of the detected TRAPPC2 mutations [7]. This unusually high deletion frequency, particularly in a gene encoding only a small protein of 140 amino acids, may possibly be explained by the 5 truncated pseudogenes located on chromosome Yq11.23 [8], which may cause homologous recombination and slipped mispairing [2]. The other reported types of TRAPPC2 gene disruptions include 9 splice-site mutations, 7 nonsense mutations, 6 missense mutations, and 2 insertion mutations [7]. Among the recurrent gene disruptions, the most frequent variations are the nucleotide transition c.93+5G>A (formerly described as IVS+5G>A) and the nucleotide deletion c.271-275delCAAGA (p.Gln-91Argfs*9). These variations have been identified in 16 unrelated families thus far, and are responsible for 28% of all analyzed cases. The G to A transition in the 5th base of the intron 3 splice donor site (c.93+5G>A), the mutation that was also identified in our case, has been shown to produce aberrant transcripts lacking exon 3; since the translation initiation site is located in exon 3, it is hypothesized that no TRAPPC2 protein is produced from this mutant allele [9].
Given the spectrum of mutations of the TRAPPC2 gene, the phenotypic findings in families with X-linked SEDT have shown a remarkable degree of homogeneity [2], and there is no clear genotype-phenotype correlation for TRAPPC2 mutations known to date. There are reports on a trend of decreasing severity, in terms of kyphosis, scoliosis, and degree of pain, in the order of the mutations from the 5' to 3' ends along the TRAPPC2 gene [2, 7]. In our case, however, the mutation was located in the 5' region of the gene, but the patient was asymptomatic with no complaints of back or hip pain, and an absence of radiological evidence of kyphosis and scoliosis. Therefore, the trend of increased severity towards the 5' end of the ORF noted in previous reports was not observed in our case.
The clinical diagnosis of X-linked SEDT is usually based on clinical and radiological features. However, the skeleton is usually normal in childhood [2], and the characteristic radiological manifestations are usually recognizable in mid- or late adolescence [10]. Therefore, in the absence of clear radiologic features, the diagnosis of X-linked SEDT rests on the recognition of the mode of inheritance [2], and molecular analysis is eventually needed for the confirmatory diagnosis of X-linked SEDT, especially in sporadic and/or atypical cases.
Herein, we present a case of X-linked SEDT, which was diagnosed by a combination of clinical and radiographic features, and pedigree analysis that was confirmed by TRAPPC2 mutation analysis. While the diagnosis of X-linked SEDT is still based on clinical and radiographic grounds, there is growing demand for the molecular characterization of causative mutations. Taken together, our report of a genetically confirmed X-linked SEDT case for the first time in Korea reveals the characteristic X-linked mode of inheritance in a large Korean pedigree and accentuates the role of molecular TRAPPC2 mutation analysis in confirming the diagnosis of X-linked SEDT.

 XML Download
XML Download