

 PDF
PDF Citation
Citation Print
Print



To the Editor:
We read with interest Joo et al.’s article1 on a prospective, single-centre, study of the frequency of six mitochondrial DNA (mtDNA) variants known to be associated with hearing loss (HL) in a cohort of 1,099 patients from 711 Korean families with pre- or post-lingual HL. It was found that 12 of the 1,099 patients carried either the m.1555A>G variant (n = 10) or the m.7444G>A variant (n = 2).1 It was concluded that a significant proportion of Korean patients with HL carried mtDNA variants with m.1555A>G being the most common, therefore there is a need for genetic testing in patients with HL.1 The study is excellent, but some points require discussion.
First, the heteroplasmy rates of the m.1555A>G and m.7444G>A variants were not determined in all patients with them.1555A>G variant.1 To assess whether there is a causal relationship between the mtDNA variants and HL, it is important to know the individual heteroplasmy rates in different tissues. In addition, mtDNA copy numbers should be specified, as these also have a significant influence on the phenotype. Homoplasmy was reported in eight patients carrying the m.1555A>G variant.1
Second, carrying an mtDNA variant associated with HL does not necessarily mean that that particular variant is responsible for HL. Several modifying factors can influence phenotypic expression, including heteroplasmy, mtDNA copy numbers, haplotype, and nuclear genes involved in mitochondrial regulation and reproduction. Did the degree of heteroplasmy correlate with the degree of HL?
Third, we disagree that the exact association between visual impairment, hypothyroidism, and diabetes with HL is unknown.1 Diabetes, hypothyroidism, and visual impairment are common phenotypic features (red flags) of mitochondrial disorders (MIDs) due to mtDNA variants and may also occur in isolation or together with other phenotypic features in multisystem MIDs. The combination of diabetes, hypothyroidism, and visual impairment even suggests that MID could be the underlying cause. Visual impairment in MIDs may be due to cortical/subcortical stroke-like lesions, optic atrophy, retinitis pigmentosa, or diabetic retinopathy.
Fourth, according to Table 3 of the index article, one patient had a history of exposure to noise (patient 379_21).1 How did the authors rule out that HL was due to the acoustic trauma and not the MID?
Fifth, there is a discrepancy between the method and the results section in the abstract. According to the method section, 1,099 patients from 711 families were included in the study. According to the results, 711 patients were examined. This discrepancy should be resolved.
Sixth, pre- and postlingual HL is particularly associated with the variants m.1555A>G and m.7444G>A, but can generally be found in any of the mtDNA mutations.2 Depending on the disease stage and modifying factors that influence the phenotypic expression of an mtDNA, variant, in principle, all syndromic and non-syndromic MIDs can be associated with HL.
Seventh, we should know why two patients with autosomal recessive inheritance Table 3 of the index article were included in the analysis. HL in these two patients cannot be due to an mtDNA variant. This discrepancy should be resolved.
We disagree with the statement that the absence of patients with HL who are carriers of the m.3243A>G variant does not necessarily mean that HL in m.3243A>G carriers is extremely rare.1 On the contrary, HL is a common feature of MELAS.
Overall, the interesting study has limitations that call into question the results and their interpretation. Clarifying these weaknesses would strengthen the conclusions and could add value to the study. HL is more prevalent in MID patients than expected. Factors affecting the phenotypic expression of pathogenic mtDNA variants need to be considered when analysing mtDNA-associated HL.
Authors' Response to the Letter
We acknowledge and value Dr. Josef Finsterer’s comprehensive correspondence regarding our article. Here, we aim to discuss the concerns and limitations brought forth by him.
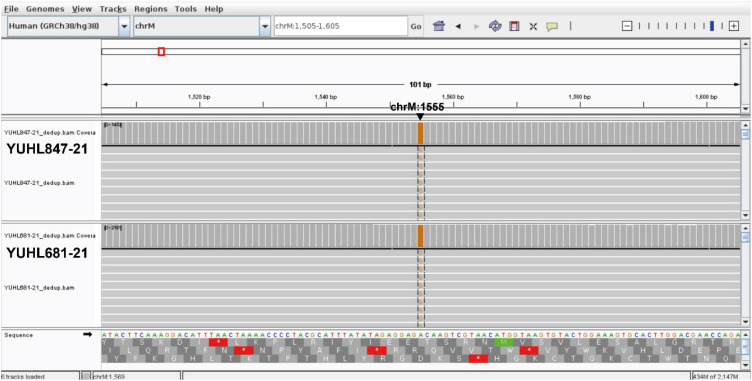
First, we acknowledge that investigating the correlation between the heteroplasmy rate of mtDNA variants and the degree of hearing loss is an intriguing aspect to examine. Nevertheless, within the confines of our study, it is difficult to accurately and precisely determine the heteroplasmy rate or mtDNA copy numbers in patients. We focused primarily on restriction fragment length polymorphism (RFLP) approach to identify pathogenic mtDNA variants. RFLP is a fast and cost-effective screening method, and the homoplasmy of mtDNA variants in eight patients was inferred from the sequence chromatogram of sequenced amplicons. However, RFLP is not sensitive enough to evaluate uneven copies of mtDNA in blood and detect low-level heteroplasmy. This limitation primarily arises from the inherent bias in the amplification bias during PCR, as described in our article.1 Mutect2, a tool provided by GATK (Genome Analysis Toolkit, https://gatk.broadinstitute.org/hc/en-us), is highly effective and sensitive in identifying low-frequency variants such as low-level heteroplasmic mtDNA variants. However, it is important to note that Mutect2 requires whole genome sequencing (WGS) data as input.3 Fortunately, we had WGS data from two out of ten individuals who had the m.1555A>G variant. Among the two patients who had WGS data, YUHL681-21 was detected with 2,022 copies of mitochondrial DNA, all of which included the identical m.1555A>G variant at the site, implicating homoplasmic m.1555A>G variant in this patient Fig. 1.1 Furthermore, YUHL847-21 was found to have a total of 1,347 mitochondria, with only one of them being a different mitochondrial allele from m.1555A>G. This suggests that the m.1555A>G variant is homoplasmic, as an alternate allele frequency above 95% from WGS data is classified as homoplasmic according to gnomAD (v4.0.0) Fig. 1.14 Due to the limited number of available patients (YUHL 681-21 and 847-21), it is currently challenging to examine the precise association between the heteroplasmy rates of affected individuals and the degree of hearing loss. Furthermore, assessing the heteroplasmy rates in various tissues was hard to perform because obtaining biopsied tissues, such as muscle, from patients with isolated hearing loss is uncommon and completely unnecessary under standard clinical care.
Second, as Dr. Finsterer stated, there are other modifying factors that affect the penetrance and expressivity of hearing loss caused by mtDNA variants. Therefore, we assessed additional variables that could potentially influence the outcome, such as the exposure to aminoglycoside, the mtDNA haplotype, the presence of mutations in modifier genes such as TRMU, and any other pathogenic or likely pathogenic mutations in hearing loss-associated nuclear genes for patients whose whole exome or genome sequencing were available. Detailed results of this evaluation are already available in our article.1
Third, Dr. Finsterer pointed out that diabetes, hypothyroidism, and visual impairment are common phenotypes of syndromic MID, alongside hearing loss. As stated in our article,1 we acknowledge that the involvements of multiple mtDNA variants in conditions such as diabetes like maternally-inherited diabetes and deafness (MIDD), visual impairment like Leber’s hereditary optic neuropathy, and hypothyroidism. However, there is a paucity of data supporting the notion that m.1555A>G or m.7444G>A is a sole cause of those phenotypes. In fact, there is no documented association between m.1555A>G or m.7444G>A variant and diabetes, visual impairment, or hypothyroidism according to OMIM (https://omim.org/) and Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/) databases. In addition, these phenotypes were not co-segregated within affected family members. Thus, while it is not entirely incorrect to hypothesize that the observed syndromic phenotypes may be caused by the identified mtDNA variants combined with unknown modifying factors, adhering to the ACMG standards and guidelines for the interpretation of sequence variants,5 we cannot ascertain that the m.1555A>G or m.7444G>A variant is responsible for the observed phenotypes, such as diabetes, visual impairment, and hypothyroidism.
Fourth, YUHL379-21 had a history of noise exposure in his twenties. However, this exposure alone was inadequate in explaining his profound hearing loss because the onset of his hearing loss occurred before the noise exposure, and the intensity and duration of the noise exposure were insufficient to cause a permanent threshold shift. Hence, it is reasonable to conclude that the m.7444G>A mtDNA variant detected in this patient is more likely a factor contributing to hearing loss.
Fifth, we apologize for the confusion that we made in the methods and results sections. In the results section, the term “patients” refers to “probands.” In order to avoid increasing the detection rate in our cohort, we selected one affected proband from each family, regardless of whether the family had multiple affected or unaffected members. There is no change in the description that we performed screening for 1,099 individuals from 711 families, including 711 probands. To enhance clarity, we would like to revise the wording in Fig. 1 as follows: “10 unrelated hearing loss probands with the m.1555A>G variant and 2 probands with the m.7444G>A variant.”
Sixth, Dr. Finsterer inquired about the inclusion of two probands (YUHL58-21 and 82-21) who exhibited autosomal recessive inheritance in our study. As evidenced by the segregation analysis presented in our article (Supplementary Fig. 1),1 the m.7444G>A variant of YUHL 58-21 is inherited maternally. This is supported by the fact that both the patient’s mother and brother also carried the same mtDNA mutation. The observed phenotype in the family may be explained by incomplete penetrance. Unfortunately, we were unable to obtain samples from other family members of YUHL82-21 in order to determine whether the detected mtDNA variant occurred sporadically or was inherited maternally from his mother with incomplete penetrance. Like these instances, incomplete penetrance of hearing loss resulting from mtDNA variants is not uncommon. This might lead to misinterpretation of the inheritance pattern based solely on the observed phenotype. Hence, we assessed the mtDNA variants linked to hearing loss in all 711 families, irrespective of the inheritance pattern. Moreover, there is a growing body of research indicating that de novo mtDNA mutations are responsible for MID.678910 Therefore, comprehensive screening of mtDNA variants in a large hearing loss cohort with various modes of inheritance and varying degrees of hearing loss is a more appropriate approach to accurately determine the prevalence of these variants.
Seventh, Dr. Finsterer expressed his disagreement with the assertion that hearing loss is exceptionally uncommon in individuals with the m.3243A>G variant. We did not dispute the rarity of hearing loss in individuals with m.3243A>G variant. Among Korean patients with hearing loss, the occurrence of m.3243A>G variant is very rare, however, it does not mean that m.3243A>G does not cause hearing loss.
Finally, we express our gratitude to Dr. Finsterer for the critical reading and insightful comments about our article. Based on this response, we believe we have thoroughly clarified the issues he raised.
1Department of Pharmacology, Graduate School of Medical Science, Brain Korea 21 Project, Yonsei University College of Medicine, Seoul, Korea
2Won-Sang Lee Institute for Hearing Loss, Seoul, Korea
3Department of Otorhinolaryngology, Yonsei University College of Medicine, Seoul, Korea
*Sun Young Joo and Seung Hyun Jang contributed equally to this work.
Address for Correspondence: Jinsei Jung, MD, PhD. Department of Otorhinolaryngology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea. jsjung@yuhs.ac
Address for Correspondence: Heon Yung Gee, MD, PhD. Department of Pharmacology, Graduate School of Medical Science, Brain Korea 21 Project, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea. hygee@yuhs.ac
 XML Download
XML Download