

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
In recent years, the demand for cell and gene therapy (CGT) has surged as the number of United States Food and Drug Administration (U.S. FDA)-approved CGT products has increased annually [1]. This increasing demand underscores the pressing need for manufacturers to improve and advance their production processes to keep pace with the rapidly evolving CGT field. The entire CGT manufacturing process must occur in a facility that complies with good manufacturing practice (GMP) regulations and has received full authorization to manufacture CGTs or advanced-therapy medicinal products [2], as they are referred to in Europe, for human use. Numerous clinical trials have been conducted to assess the safety and efficacy of CGTs for a wide range of clinical indications, including cancer [3-5]. CGT includes the injection of cells, genes, or gene-modified cells into a patient with the purpose of preventing or treating a disease. Cell therapy involves the injection of cells to replace or aid the repair of damaged cells, whereas gene therapy involves the injection of genes into cells to alter the genetic makeup of the recipient cells [1, 6, 7]. Cell-based gene therapies, such as chimeric antigen receptor T (CAR-T) therapy, represent combinations of both approaches.
According to a recent survey of CGT investors conducted by Bloomberg Intelligence and the Commercialization Committee of the International Society for Cell & Gene Therapy, clinically significant data are the number one consideration for investors in the CGT field [8]. Maintaining the efficacy of a cell and/or gene therapy product during the manufacturing process is a key factor that can influence overall clinical outcomes. Challenges faced when manufacturing therapeutic products must be addressed to ensure their efficacious clinical translation. In this review, we provide a general overview of representative CGTs, namely mesenchymal stem cells (MSCs) for cell therapy, lentiviral vectors (LVs) and adeno-associated viral vectors (AAVs) for gene therapy, and CAR-T cells for cell-based gene therapy. We also summarize the general CGT manufacturing processes and highlight challenges encountered during manufacturing that may potentially hinder the clinical use of the respective CGTs.
MSC-BASED CELL THERAPIES
Various cell types, including T cells, stem cells, dendritic cells, and natural killer (NK) cells, have been investigated in clinical trials involving MSC-based cell therapies [9]. Cancer is the main indication for leukocyte (T cells, dendritic cells, and NK cells) therapy. Following leukocytes, the second most commonly used type of cell therapy is stem cell-based therapy, which involves hematopoietic stem cells (HSCs) or MSCs [10]. Although numerous autologous stem cell transplantations have been performed for MSC therapy, allogeneic cells have been predominantly used for that purpose [11, 12]. MSCs are readily available and can be acquired from various sources, including the bone marrow, adipose tissue, and umbilical cord tissue [13, 14]. The mechanism of action of MSCs has been attributed to paracrine activity rather than the direct replacement of damaged cells [15]. Paracrine activity refers to the ability of MSCs to secrete paracrine factors into the microenvironment [16].
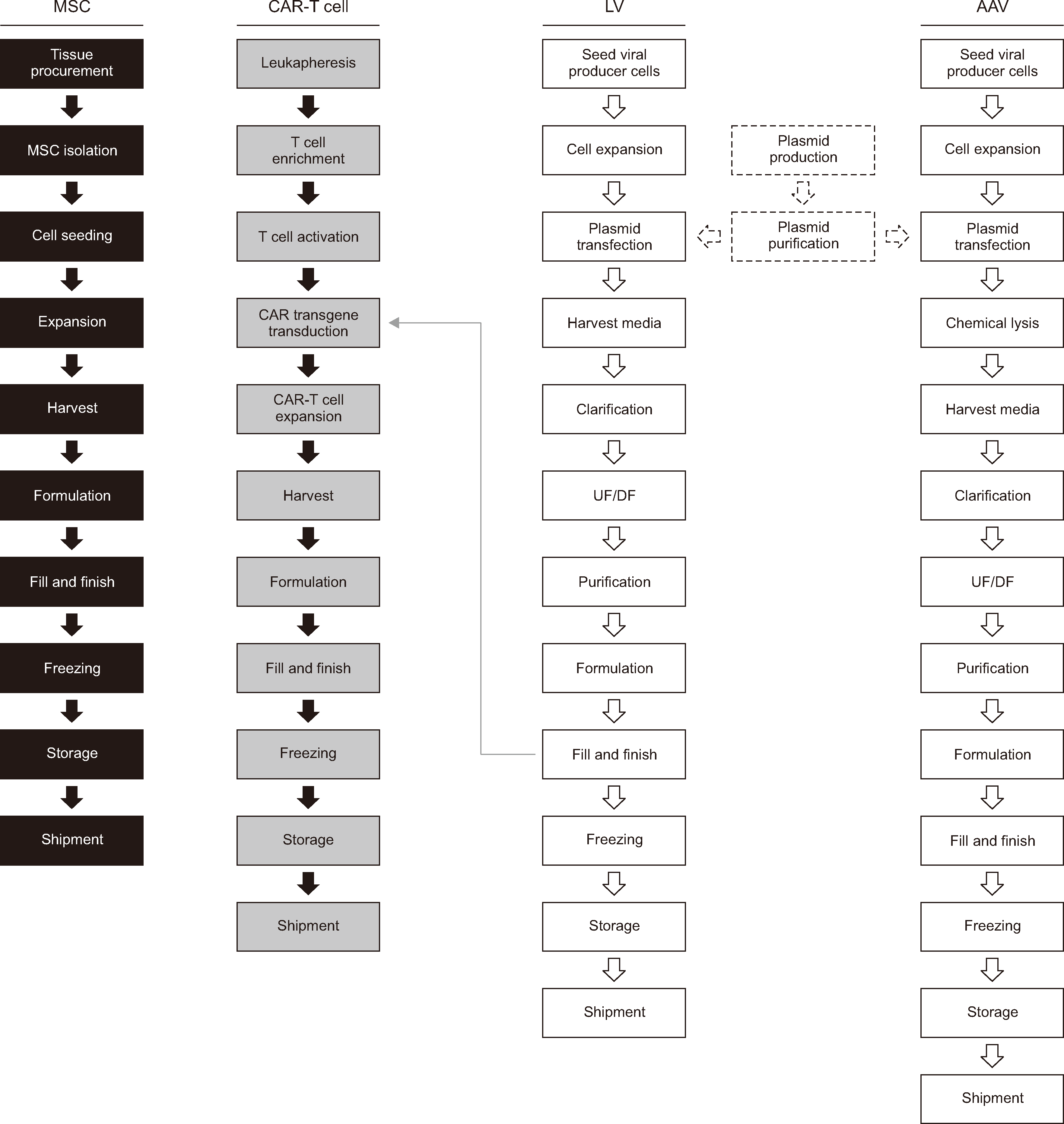
The upstream MSC manufacturing process (Fig. 1) starts with tissue procurement, followed by MSC isolation from the tissue source. Different techniques are employed to isolate MSCs from tissues, depending on the tissue source [14]. Cells are cultured in a conventional culture medium that usually contains supplements, such as fetal bovine serum (FBS). The MSCs are then expanded on different scales, ranging from multilayer flasks to bioreactors [17, 18]. The duration of cultivation and number of passages differ among manufacturers and institutions. The identity, potency, and safety of the cells are confirmed via QC analysis. The minimum criteria for characterizing MSCs include (but are not limited to) their identity, sterility, viability, purity, potency, and efficacy [19]. To prepare and distribute cells for injection, they are harvested after expansion and subjected to the following steps: formulation, fill and finish, freezing, storage, and shipment.
Common challenges faced in MSC manufacturing are presented in Table 1. To achieve clinical doses, MSCs must be expanded extensively. However, during expansion, variabilities may arise depending on the donor, the condition/sterility of the tissue source or starting material, the isolation technique, and the cultivation method. Such factors influence MSC heterogeneity and potency. Sequential passaging and long-term culture of MSCs may also affect their potency, specifically their proliferation capabilities [20]. Prolonging the MSC culture duration implies the use of more culture mediums and supplements, such as FBS, raising the overall manufacturing cost. Moreover, increased exposure to FBS can amplify pre-existing safety concerns, including adverse immunological reactions elicited in recipients and the high batch-to-batch variability of FBS [17, 18]. To ensure the advancement of MSC-based cell therapies in clinical settings, it is critical to employ methods that enable rapid, large-scale MSC expansion, balancing the therapeutic potency of the cells with acceptable manufacturing expenses.
AAV AND LV-BASED GENE THERAPIES
AAVs and LVs are important viral vectors used for gene therapies [21]. Currently, FDA-approved, commercially available AAV-based therapies target diseases such as retinal dystrophy and spinal atrophy [22], whereas LV-based gene therapies target diseases such as B-cell lymphoma, β-thalassemia, and cerebral adrenoleukodystrophy [23]. Both vectors package genes and enable direct administration of the gene of interest to patients [24]. A major difference between the two types of vectors is that a single DNA strand is packaged in AAVs, whereas an RNA genome is packaged in LVs. The advantages of AAVs include low immunogenicity, high delivery and efficiency, and long-term expression. The advantages of LVs include long-term transgene expression and the ability to transduce nondividing cells [25]. LVs permanently integrate into the host genome. Although the overall frequency is low, genomic integration is also possible for AAVs [26, 27].
A plasmid-based approach is commonly used to manufacture AAVs. The upstream AAV manufacturing process (Fig. 1) [28] starts with large-scale plasmid production and purification. The following plasmids must be prepared for transfection: (1) a cis-acting plasmid that carries the therapeutic gene of interest, (2) a trans-acting plasmid containing protein-coding genes necessary for AAV replication and capsid formation, and (3) a trans-acting plasmid that enables AAV replication in the host cells [29]. All three plasmids are transfected into an AAV-producing cell line, such as human embryonic kidney 293 (HEK293) cells, to produce viral vectors. Cell stocks stored in a cell bank are thawed and cultured before large-scale expansion (growth in single-use bioreactors). Following chemical lysis, which involves the disruption of cells to release AAVs into the culture medium, nuclease treatment is performed to purify the vectors by selectively degrading DNA impurities. Subsequently, the harvested viral supernatant undergoes a purification process comprising centrifugation, chromatography, and ultrafiltration (UF) and diafiltration (DF) to remove cellular debris and impurities. The impurities consist of plasmid DNA, proteins and DNA from AAV-producing cells, and empty capsids.
QC assays must be performed to assess the identity, potency, and safety of the viral vectors. Examples of QC tests include the detection of viral contamination, residual DNA, and residual proteins, as well as the measurement of the vector genome concentration [30]. The final manufacturing steps include formulation followed by fill and finish, freezing, storage, and shipment. Processing, filtration, and/or centrifugation methods are implemented to remove impurities from the viral supernatant. The final formulation of LVs is achieved during the final fill-and-finish step. As for AAVs, QC requirements have been established for LVs and include tests such as assessing their physical and functional titers and detecting and removing impurities [31].
The upstream LV manufacturing process (Fig. 1) [32] starts with the culture and large-scale expansion (i.e., in a bioreactor) of an LV-producer cell line, such as HEK293T, a derivative of the HEK293 cell line that expresses the SV40 T-antigen. SV40 T-antigen expression prevents innate immune response activation. HEK293T cells can also be used to produce LVs at high titers [33]. However, the presence of the SV40 T-antigen raises safety concerns regarding clinical manufacturing [34]. Therefore, LV-producing cells that do not express the SV40 T-antigen are being developed for clinical purposes. Furthermore, because of concerns regarding the use of xenogeneic serum (FBS) and the removal of residual serum during purification, well-established methods have been implemented to culture LVs in serum-free medium that yields titers comparable to those obtained with serum-supplemented culture medium [33]. Plasmids encoding the transgene of interest and the vesicular stomatitis virus G protein, envelope, gag, and rev genes can be transiently expressed in HEK293T cells using a transfection reagent. Usually, the cells are co-transfected with three to four plasmids. Unlike AAVs, most LV vectors are released from the cells, and the culture supernatant is used for the downstream process [32].
Although commonly used, transient expression may not be ideally suited for large-scale manufacturing of AAVs and LVs because of scalability issues. Large amounts of transfection reagents and genetic material (DNA) are required to scale up transient expression systems, which leads to increased production costs and, potentially, batch-to-batch variability [35]. An alternative option is to establish and use stable producer cell lines. Stable producer cell line production requires the use of helper viruses, such as an adenovirus [36]. However, it does not require plasmid DNA because the genes required for viral vector production are already integrated into the cell lines. Using stable cell lines results in fewer empty capsids and better vector quality than transient expression [37]. Using stable producer cell lines can also reduce manufacturing costs and maximize the titers of both AAVs and LVs [36, 38]. Therefore, employing stable producer cell lines can help overcome challenges currently faced in gene therapy, such as a low product yield.
Challenges faced in AAV and LV manufacturing are presented in Table 1. Purification is a major hurdle for both AAVs and LVs. The purity of AAVs and LVs varies depending on the vector-biomanufacturing process, and residual impurities may affect the final product yield. Developing an effective, scalable purification method is crucial to reduce the overall cost of GMP-compliant production and increase vector production [39]. Empty-full separation (i.e., the separation of empty and partial capsids from full AAV capsids) is another challenge that may affect the clinical use of AAV-based therapies. Contamination with empty AAV capsids may generate immunological reactions in the host, such as enhanced T-cell proliferation [40]. Transfection conditions must be optimized to maximize AAV and LV production. Although the use of stable producer cell lines may serve clinically beneficial purposes, several points must be considered during manufacturing to facilitate clinical translation, such as contamination with helper viruses or cytotoxicity induced by vector components required for AAV production [41]. The purification process is another critical step in LV manufacturing because impurities (such as residual DNA, HEK293T cell proteins, medium components, and transfection reagents) can generate unwanted inflammatory responses. The inherently unstable nature of LVs [42] is a major challenge during production, in which environmental factors can influence and abrogate LV functionality [43].
CAR-T CELL-BASED GENE THERAPIES
Both cell and gene manufacturing are required for the production of CAR-T cells (Fig. 1). Predominantly used in cancer immunotherapy as a treatment option for B-cell lymphoma and leukemia, CAR-T cell therapy involves genetically modifying T cells ex vivo, which enables cancer-cell targeting and elimination after CAR-T cells are infused into the patient. T cells can be genetically engineered to recognize B-cell maturation antigen (BCMA) [44] or B-cell surface antigens, such as CD19 and CD20 [45, 46]. To evade on-target toxicity in normal cells, CAR-T cell targets should be highly selective [47].
CAR-T cell manufacturing [48, 49] starts with leukapheresis, i.e., the collection of leukocytes from the blood of the patient (autologous) or a healthy donor (allogeneic). The leukapheresis material is then processed to enrich and isolate T cells. Viral vectors, usually LVs or retroviral vectors, must be prepared along with the T cells to transduce the latter with the CAR transgene. With respect to clinical applications, LVs are attractive candidates because they can transduce slowly proliferating or non-proliferating cells more efficiently than other vectors [50]. Antigen-presenting cells (such as dendritic cells) are used to activate T cells in vivo, facilitating their stimulation for proliferation and differentiation [48]. For the purposes of clinical manufacturing, the most common approach to activate T cells involves the use of a monoclonal antibody (such as an anti-CD3 antibody) and an interleukin (such as interleukin-2) [48]. Activated T cells are transduced with the CAR transgene via LVs and expanded in culture vessels. The use of serum in the culture medium must be contemplated. Previous reports have shown that serum can affect T-cell functionality [51] and that using serum-free medium enhances the killing potential of both T cells and CAR-T cells in vitro and in vivo [52]. After formulation and filling, the final steps of CAR-T cell manufacturing include freezing, storage, and shipment.
The quality of the starting leukapheresis material must be scrutinized to increase the efficacy and potency of CAR-T cell therapy during the clinical stage. For CAR-T cell therapy, the purity and potency of both the isolated T cells and the LVs must be considered. The technique used to select and isolate T cells must be optimized to ensure that a pure T cell population is used as the starting material. Contaminating monocytes remaining in the starting material disrupt T-cell activation and transduction, potentially resulting in poor T-cell quality and CAR-T cell manufacturing failure [53]. In addition to confirming the purity of the T cells, QC requirements for CAR-T cells include examining cell-surface CAR expression and sterility testing (i.e., potency assays), which must be conducted quickly as CAR-T cells are generally infused shortly after production.
The time between T-cell collection and CAR-T cell infusion (i.e., the vein-to-vein time) must be considered during manufacturing. Vein-to-vein time optimization is crucial for patients in need of urgent treatment. The vein-to-vein time, including the time required for manufacturing and quality assessment, reportedly is three to five weeks [54]. Concerns of deterioration of the patient’s condition during the interim and adverse disease progression are always present. CAR-T cell manufacturing is shifting toward the use of a closed, automated, single-use GMP-compliant system that can yield a CAR-T cell product within two weeks [55, 56]. This advancement will provide a standardized method for producing CAR-T cells and eliminate the influence of manual handling or manipulations on the overall quality of the manufactured CAR-T cell product.
QC TESTING
As mentioned earlier, the overall manufacturing process for CGTs is arduous and complicated. Performing proper in-process QC and validation tests is of utmost importance for translating the respective therapeutic products into clinical use. Several attributes, such as identity, purity, potency, and safety (sterility and the presence of adventitious agents, endotoxins, or mycoplasma), are commonly examined for different CGTs. Table 2 lists various tests and measurement methods used to assess quality attributes [19,30,31,57,58]. Considering the importance of time for CAR-T cell therapies, efficient and rapid QC tests should be implemented in manufacturing. Nucleic acid amplification tests that can detect mycoplasma infections more rapidly than conventional methods are currently available. Demonstrating that a viral vector is not contaminated with replication-competent vectors (RCVs) is essential for related therapies because RCVs may affect the behavior of the viral vector [59].
Each manufacturer has specifications that must be met to release a finished product. Publicly disclosed lot-release specifications for four representative FDA-approved CAR-T cell products, namely Kymriah [60], Yescarta [61], Tecartus [62], and Carvykti [63], are presented in Table 3. Kymriah, Yescarta, and Tecartus target CD19, whereas Carvykti targets BCMA. Although Kymriah targets CD19 and Carvykti targets BCMA, the presence of CARs is verified for both products to confirm their identity. Potency testing for Kymriah focuses on interferon-gamma release, whereas Carvykti testing focuses on CAR expression in viable T cells, along with a non-disclosed component. Although Kymriah, Yescarta, and Tecartus all target CD19, different methods are used to evaluate their quality and safety before the final product release. Major distinctions exist regarding purity assessments. Purity specifications for Tecartus are unavailable, preventing a direct comparison with the other CD19-targeting CAR-T cell therapies. However, the evaluation methods used for Kymriah and Yescarta differ markedly. Gentamicin, endotoxin, and a non-disclosed reagent are assessed when testing the purity of Yescarta, whereas the percentage of viable T cells or CD19-positive B cells is quantified for Kymriah.
The variable methods used to evaluate the quality and safety of different FDA-approved CAR-T cell therapies underscore the necessity for a standardized testing approach. The U.S. FDA’s draft guidance for the industry on developing CAR-T cell therapies [64] offers potentially valuable insights into assay development and evaluation methods for critical quality attributes, including identity and potency. Regarding identity, the recommendations advocate using flow cytometry or PCR to detect the transgene. The document suggests employing cell-surface markers to observe the cellular composition of the final products and emphasizes testing the potency of both vectors and CAR-T cells [64]. It also strongly recommends using a matrix approach involving various assays, such as cell-killing assays, cytokine-secretion assays, and transduction-efficiency measurements to confirm the potency.
FUTURE PERSPECTIVES
Determining whether centralized or decentralized manufacturing is a more appropriate manufacturing model is a subject of debate in the CGT field [65-67]. Centralized manufacturing involves the manufacturing of products at a centralized GMP facility and the distribution of the products to point-of-care locations. With decentralized manufacturing, local production is possible when the GMP facility is close to the point-of-care location, which substantially reduces the time required to deliver the products. Applying the decentralized model to cell-based therapies, such as CAR-T cell therapy, will be advantageous, considering that rapid product delivery to patients with cancer is essential. Facilitating multi-center manufacturing, however, would require standardizing the manufacturing protocol to minimize product variation.
To successfully deliver the manufactured products for clinical use, patient accessibility must be prioritized, regardless of which manufacturing model is implemented. In-house manufacturing in a GMP facility of a hospital [68] represents a centralized approach to manufacturing and secures patient accessibility. In-house manufacturing within the hospital can help overcome many challenges faced when manufacturing CGTs for clinical use. The quality of the tissue source (starting material) can be ensured through rapid delivery from the operation room to the GMP facility, which would reduce the time delay between manufacturing and clinical application and enable parallel production and patient monitoring at a single location.
These features of in-house manufacturing within the hospital setting will benefit allogeneic and autologous therapies involving both stem cell and CAR-T cell therapies. If a hospital has no GMP facility, CAR-T cell production must be outsourced, involving local cell collection, cryopreservation, and shipping to the manufacturing site. Outsourcing of the manufacturing process (including QC), followed by the shipment of the final product back to the point-of-care location, increases the vein-to-vein time. Real-world analysis has indicated that the vein-to-vein time of representative, currently available CAR-T cell products is longer than 28 days [69]. In-house GMP manufacturing within the hospital may serve as a solution to reduce the vein-to-vein time.
The high cost and GMP regulatory compliance are just a few of the multiple challenges hospitals face in building a GMP facility. Hospitals must be able to pay for the costs of building and maintaining a manufacturing facility, and the facility must abide by GMP laws and regulations [68]. If hospitals cannot overcome obstacles associated with building an in-house GMP facility, an alternative would be for them to start a spin-off company. These spin-off firms could maintain a strong network with local hospitals and specialize in meeting their manufacturing needs. Ideally, the spin-off companies would provide one-stop shopping services where the production and manufacturing of cell, gene, and cell-based gene therapies occur at a single location near the point-of-care location and, most importantly, the patient.
CONCLUSION
CGT is a pertinent topic because it represents a promising strategy for treating a broad spectrum of diseases. The increasing attention to CGTs highlights the complexities of the manufacturing process, which can impede the clinical application of the manufactured products. To progress from demonstrating potential to showcasing robust clinical efficacy, addressing and overcoming the manufacturing challenges associated with CGTs is imperative.
 XML Download
XML Download