

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hyperthyroidism is mainly characterized by excessive synthesis and release of thyroid hormones (THs) by the thyroid gland, causing hyper-metabolism in the body [1,2]. Hyperthyroidism-induced cardiomyopathy (HTC), one of the most common complications of hyperthyroidism, is harmful to the cardiac conduction system and cardiomyocytes caused by the direct or indirect toxic effects of chronic excess THs on the heart [3,4]. It is the main cause of death in hyperthyroidism other than hyperthyroid crisis [5]. In contrast, myocardial fibrosis is a key factor affecting the progression of HTC. THs induce endoplasmic reticulum stress (ERS) [6] and apoptosis in cardiomyocytes [7] while increasing the force and speed of myocardial contraction [8] and the burden on the heart. Thus, it can lead to cardiac hypertrophy and left ventricular dysfunction [9], resulting in heart failure [10,11] and dilated cardiomyopathy [12]. Currently, the mechanism of hyperthyroidism-induced myocardial fibrosis remains unclear, and there are few studies on clear and effective clinical interventions for hyperthyroidism-induced myocardial fibrosis.
Apoptosis is a unique and important mode of “programmed” cell death that involves the genetically determined cell elimination [13]. Hyperthyroidism can induce apoptosis in liver cells [14] and myocardium of hyperthyroid rats [7], but the specific intrinsic regulatory mechanisms are not clear. Homeostatic imbalance of the endoplasmic reticulum (ER), an important organelle in eukaryotic cells, will lead to ERS [15], which is closely associated with several cardiovascular diseases, such as myocardial infarction, myocardial ischemia, dilated cardiomyopathy, and heart failure [16]. As an ERS protein, C/EBP homologous protein (CHOP) can inhibit the expression of anti-apoptotic proteins, activate the apoptotic proteins, and initiate the Caspase signaling pathway to induce apoptosis [17]. In addition, excessive ERS can induce apoptosis [18]. Whether ERS can mediate the hyperthyroidism-induced myocardial fibrosis is still unclear and needs to be further investigated.
The Hippo signaling pathway, which plays a crucial role in regulating cellular homeostasis and is a significant mediator of apoptosis, represents a promising target for intervention in various many cardiovascular diseases [19]. Blocking the Hippo pathway has been demonstrated to reduce apoptosis and thus counteract fibrosis [20]. Specifically, the activation of Yes-associated protein (YAP) is a critical component of Hippo signal transduction, which is essential for apoptosis during ERS [21]. Furthermore, recent research suggests that downregulation of the Hippo pathway could ameliorate myocardial injury by suppressing ERS [22,23]. However, the precise contribution of the Hippo pathway-mediated apoptosis to the development of HTC remains unclear.
Sulfur dioxide (SO2), once considered a toxic gas with an unpleasant odor, is now recognized as a novel endogenous gas signaling molecule skin to nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S). In mammals, SO2 is produced endogenously through the transamination of aspartate aminotransferase (AAT) during the metabolism of sulfur-containing amino acids [24]. Studies have demonstrated that SO₂ can ameliorate myocardial fibrosis caused by myocardial infarction or diabetes by inhibiting the proliferation and migration of cardiac fibroblasts [25]. Nonetheless, it remains unclear whether SO2 can attenuate hyperthyroidism-induced myocardial fibrosis and whether endogenous SO2 is involved in the regulatory mechanisms of ERS and apoptosis in hyperthyroid rat cardiomyocytes.
Thus, this study aims to establish an animal model of hyperthyroidism to observe the changes in myocardial interstitial fibrosis and investigate the effects of exogenous SO2 donor (Na2SO3/NaHSO3 solution) and SO2-producing enzyme inhibitor (hydrogen deuterium exchange [HDX]) on myocardial fibrosis in hyperthyroid rats. Meanwhile, it intends to explore whether the underlying mechanisms of SO2 involve downregulating Hippo pathway to antagonize ERS and inhibit apoptosis. The findings of this study are expected to yield novel intervention targets and therapeutic approaches for the prevention and treatment of HTC.
METHODS
Animals
Animal experiments were conducted in accordance with the Guidelines for the Welfare and Use of Experimental Animals and the animal management regulations and specifications of the University of South China Laboratory Animal Center. Meanwhile, they were approved by the Animal Ethics Committee of the University of South China and conformed (Granted ID: USC201912SD23).
Study design
Forty male Sprague Dawley (SD) rats weighing (200 ± 20) g at 8 weeks of age were firstly acclimatized and fed for 1 week. After that, they were divided into 4 groups with equal number: a control group, a levothyroxine (L-Thy) group, a L-Thy + SO2 group, and a L-Thy + SO2 + HDX group. On a normal diet, L-Thy (0.5 mg/kg, Sigma-Aldrich) was intraperitoneally injected for rats in all groups except the control group [26]. Rats in the control group were intraperitoneally injected with the same dose of solvent as the model group. Both the L-Thy + SO2 and L-Thy + SO2 + HDX groups were administered intraperitoneally with SO2 donor (Na2SO3/NaHSO3, 85 mg/kg/d, Sigma-Aldrich) [27]. Furthermore, those in the L-Thy + SO2 + HDX group were additionally injected with SO2 production enzyme inhibitor (HDX, 25 mg/kg, Sigma-Aldrich) intraperitoneal with once a week for continuous 5 weeks [28].
Materials
L-Thy, Na2SO3, NaHSO3, and HDX (serine racemase inhibitor) purchased from Sigma-Aldrich. AAT1 (14886-1-AP), AAT2 (14800-1-AP), CHOP (15204-1-AP), Caspase3 (19677-1-AP), Caspase9 (10380-1-AP), Bax (50599-2-Ig), Bcl2 (26593-1-AP), MMP2 (10373-2-AP), MMP3 (66338-1-Ig), Collagen I (14695-1-AP), Collagen III (22734-1-AP), YAP1 (13584-1-AP) and GAPDH (10494-1-AP) primary antibodies and mouse/rabbit secondary antibodies were purchased from Proteintech. MST1 (3682T), GRP78/BIP (3177P), ERP72 (5033P), and Phosphor-YAP (P-YAP) (13008S) primary antibodies were purchased from Cell Signaling Technology; TIMP2 (A1558) and LATS1 (A17992) primary antibodies were purchased from ABclonal. Meanwhile, gel configuration kits and BCA protein quantification kits were purchased from Beyotime Institute of Biotechnology. Electrochemiluminescence (ECL) kit was purchased from New Cell and Molecular Biotech Co, Ltd. Total-triiodothyronine (TT3) and total thyroxine (TT4) ELISA kits were purchased from Wuhan Purity Biotech Co, Ltd. SO2 ELISA kit was purchased from Shanghai Enzyme Link Biotech Co, Ltd. Scrambled control siRNA oligonucleotide (si-NC) or siRNA for YAP1 were purchased from Hanbio Tech.
Cell culture and transfection
H9c2 cardiomyocyte cell lines, from ATCC were incubated in Dulbecco’s modified Eagle’s medium containing 5.5 mM D-glucose (10% fetal bovine serum and 1% penicillin/streptomycin) and 5% CO2 at 37°C. When the density was approximate 1 × 106, the cells were inoculated in 6-well plates. The cells were divided into a control group, a L-Thy group, a L-Thy + SO2 group, a L-Thy + SO2 + HDX group, and a SO2 group according to different treatments. The cells in the control group and the SO2 group were cultured with a normal medium, and those in the L-Thy group and L-Thy + SO2 group were treated with 2 μM L-Thy at 37°C for 24 h [29]. The cells in the L-Thy + SO2 and L-Thy + SO2 + HDX groups were pretreated for 0.5 h by adding exogenous SO2 donor Na2SO3/NaHSO3 (100 μM, 3:1 mol rate), as described previously [30]. Next, they were co-incubated with a normal medium. In contrast, the cells in the L-Thy + SO2 + HDX group were pretreated with the endogenous SO2 produced enzyme inhibitor HDX (200 μM) for 24 h [31].
In order to knock down YAP1 expression, YAP1-specific siRNA was mixed with the transfection reagent and incubated at room temperature for 15 min, as per manufacturer’s instructions. The culture medium was removed, and the cells were washed once before adding fresh antibiotic-free culture medium. The pre-mixed siRNA and transfection reagent were then introduced to the respective cell groups. The cells were then incubated at 37°C in a 5% CO2 atmosphere for 24 h. After 24 h, the culture medium was replaced to remove the siRNA and transfection reagent, and the cells continued to be cultured. Another 24 h later, cellular protein samples were extracted, and Western blot technique was employed to detect protein expression levels, thereby verifying the transfection efficiency. After successful transfection, subsequent experiments were continued.
Echocardiography
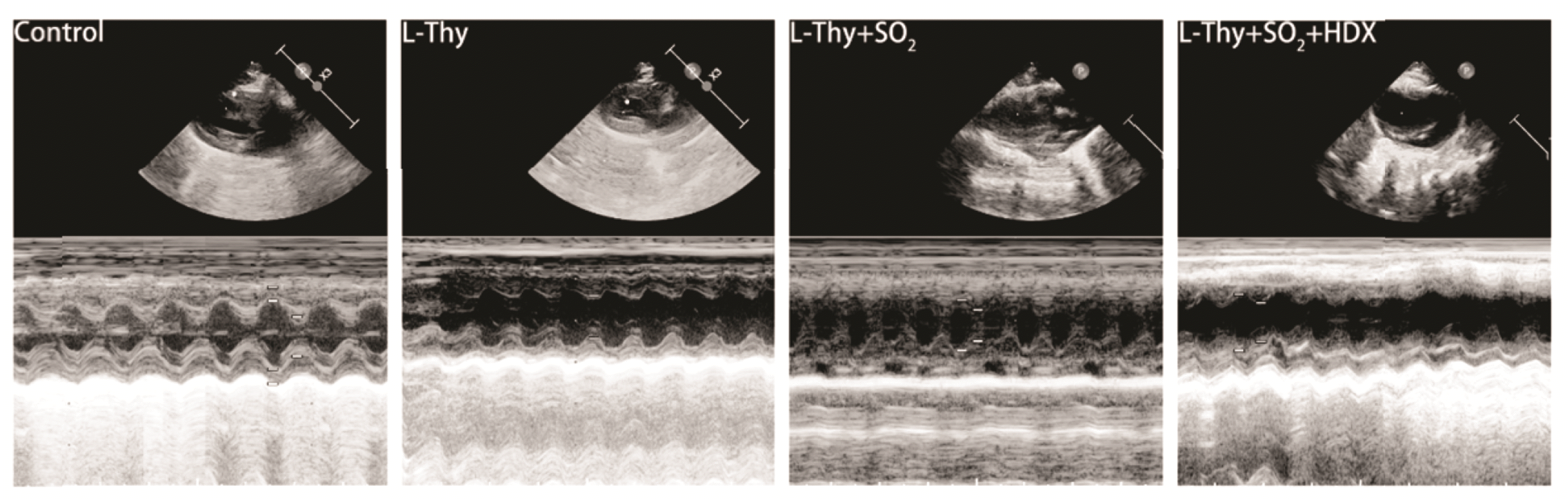
At the end of the drug intervention period, the rats were anaesthetized with 2.5% isoflurane (RWD Life Science, R510-22-10), followed by an anaesthetic respiratory system (1.5% isoflurane to maintain anaesthesia), a M-mode ultrasound machine (GE Versana Premier) and L8-18i-RS ultrahigh-frequency line-array probe were performed to measure heart rate (HR), left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) from the left parasternal axis using two-dimensional echocardiography for five consecutive cardiac cycles.
Serum TT3 and TT4 levels
Before euthanizing the rats, 2 ml of blood was collected from the renal vein and allowed to coagulate at room temperature. The blood was then centrifuged at 4°C and 3,000 r/min for 10 min to separate the serum. The serum was stored in a –80°C refrigerator until measurement. TT3 and TT4 levels in serum were quantified using the TT3 Elisa kit (CD28783; Chundubio) and TT4 Elisa kit (CD28784; Chundubio), respectively, following the instructions provided in the kit.
Calculation of heart weight (HW) index
First, the rats were weighed individually prior to the experiment. After anesthesia, the rats were placed on the operating table in a supine position and secured in place. Next, the hearts were dissected quickly and immediately rinsed with low-temperature saline to remove all blood and impurities. The hearts were then blotted with filter paper to remove excess moisture and weighed using an electronic balance to calculate the HW index. The specimens were then collected according to the required procedures.
SO2 content assay
The myocardial tissue was mashed with the appropriate amount of saline and then centrifugated. The supernatant was collected and assayed with the rat SO2 Elisa kit (YJ832161; Mlbio) according to the instruction manual.
Masson staining
The myocardial tissues were fixed using 4% paraformaldehyde, dehydrated by gradient alcohol, and embedded in paraffin. The tissues were then sectioned into 4-um-thin sections by a microtome, stained using a Masson staining kit (Abiowell Biotech Co, Ltd) according to the manufacturer’s instructions. Next, the tissues were covered with a coverslip using mounting medium. Finally, the stained tissues were examined under a light microscope.
Transmission electron microscopy (TEM)
Myocardial tissues were fixed with 2.5% glutaraldehyde, segmented into 50–100 nm thin slices, and rinsed with phosphoric acid (Beyotime Institute of Biotechnology). After fixed immersion in 1% osmium tetroxide (Absin Biosciences, Inc), the tissues were rinsed with phosphoric acid again, followed by gradient acetone immersion, dehydration, and drying. Then, they were stained with 3% uranyl acetate and lead nitrate for 10–20 min and rinsed with distilled water. Finally, they were observed under TEM to study their ultrastructure.
Western blotting
Rat myocardial tissues and cell precipitates were lysed using cold radioimmunoprecipitation assay lysis buffer containing protease inhibitors. After centrifugation, the supernatant was collected, and the protein concentration was quantified and normalized using the BCA kit, following the manufacturer’s instructions. The samples were denatured by heating at 95°C for 10 min and separated on 10%–12% polyacrylamide electrophoresis gels. The proteins were then transferred electrophoretically onto a polyvinylidene fluoride membrane (Millipore). The membranes were blocked with 5% skim milk at room temperature for 1 h and then incubated overnight at 4°C with the primary antibody. Next, they were washed three times (5 min × 3) with tris-buffered saline containing Tween 20 (TBST) and incubated with the corresponding horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. After washing with TBST (5 min × 3), the membrane was exposed to ECL chemiluminescent solution, and the image signals were acquired using the BIO-RAD XRS+ imaging system. Finally, the relative data were analyzed using ImageJ software.
Immunofluorescence staining
Cells were fixed with 4% perhalooxy-alkanes for 40 min, permeabilized with 0.1% Triton X-100 for 5 min × 2, and blocked with 5% BSA for 1 h. Next, they were incubated with rabbit anti-Bax (1:100) at 4°C overnight and then with secondary antibody coupled with FITC (1:200) at room temperature for 1 h. After that, the cells were incubated with rabbit anti-Caspase3 (1:100) at 4°C overnight and with a secondary antibody coupled with Cy3 (1:200) at room temperature for 1 h. Finally, they were re-stained using DAPI. The fluorescence was analyzed using a fluorescence microscope (Olympus IX71; Olympus).
Statistical analysis
Values are presented as the mean ± standard deviation for data that were normally distributed or median and interquartile range for data that were not normally distributed for continuous variables and number (%) for categorical variables. The representative image for each group was selected based on the mean value after quantitative analysis. Statistical analyses were performed with GraphPad Prism software (version 9.5.1). For comparisons of two groups, Student’s t-test was used. For comparisons of more than two groups, one-way analysis of variance was used, with Bonferroni’s or Dunnett’s multiple-comparisons test for normally distributed data or the Mann–Whitney test for non-normally distributed data. Unless otherwise stated, p < 0.05 was considered statistically significant.
RESULTS
TT3 and TT4 levels in the serum of rats in each group
The TT3 and TT4 levels in the serum of rats in each group were detected by the Elisa method (Table 1). Compared with the control group, the TT3 and TT4 levels in the L-Thy, L-Thy + SO2, and L-Thy + SO2 + HDX groups were significantly increased (p < 0.05). However, there was no significant differences in TT3 and TT4 levels among the three groups (p > 0.05), indicating the successful modeling of hyperthyroid rats.
Changes in SO2 content and SO2-producing enzyme (AAT1/2) expression in myocardial tissues of rats in each group
In this experiment, the SO2 contents in the myocardial tissues of rats in each group were detected by the Elisa method (Fig. 1A). The results showed that the SO2 content in the L-Thy group was significantly lower than that in the control group (p < 0.05). Additionally, compared with the L-Thy group, the SO2 content in the L-Thy + SO2 group was significantly increased (p < 0.05). Furthermore, the SO2 content in the L-Thy + SO2 + HDX group was significantly decreased compared with that in the L-Thy + SO2 group (p < 0.05). In addition, the changes in expressions of AAT1/2 in the myocardial tissues of rats in each group were measured using Western-Blot (Fig. 1B–D). Compared with the control group, the expressions of AAT1/2 in the myocardial tissues of rats in the L-Thy group were significantly lower (p < 0.05), whereas those in the L-Thy + SO2 group were upregulated (p < 0.05) after the intervention of exogenous SO2 donor (Na2SO3/NaHSO3). Besides, expressions of AAT1/2 in the myocardial tissues of rats in the L-Thy + SO2 + HDX group were significantly lower than those in the L-Thy + SO2 group after intervention with HDX (p < 0.05).
The results from in vitro cellular experiments (Fig. 1E–G) were consistent with those from the in vivo findings. The expressions of AAT1/2 were significantly decreased in H9c2 cardiomyocytes in the L-Thy group compared to those in the control group (p < 0.05). In contrast, the expressions of AAT1/2 were significantly upregulated in the L-Thy + SO2 group compared to those in the L-Thy group (p < 0.05). Moreover, H9c2 cardiomyocytes in the L-Thy + SO2 + HDX group exhibited significantly lower AAT1/2 expressions after intervention with HDX compared with those in the L-Thy + SO2 group (p < 0.05). The above results suggest that high thyroxine stimulation reduces endogenous SO2 production in myocardial tissues of hyperthyroid rats, whereas the administration of exogenous SO2 donor can increase the SO2 content and the AAT1/2 expressions in myocardial tissues of hyperthyroid rats.
Effect of SO2 on general condition and heart weight index (HW/BW) of hyperthyroid rats
The values of body weight (BW), HW, and HW/BW of rats in each group are presented in Table 2. The results indicated that the rats in the L-Thy group had significantly lower BW values and increased HW/BW values compared to those in the control group (p < 0.05). However, the rats in the L-Thy + SO2 group showed a significant increase in BW values compared with the L-Thy group (p < 0.05), while the HW/BW values demonstrated a decreasing trend without statistically significant differences (p > 0.05). Furthermore, compared with the L-Thy + SO2 group, the BW values of rats in the L-Thy + SO2 + HDX group decreased (p < 0.05), while the HW/BW values showed an increasing trend without statistically significant differences (p > 0.05).
Effect of SO2 on cardiac function in hyperthyroid rats
The hyperthyroid rats presented significant left ventricular dysfunction, which was revealed by echocardiographic analysis after 5 weeks of continuous intraperitoneal injection of L-Thy (Fig. 2, Table 3). Rats in the L-Thy group showed faster HR and decreased LVFS and LVEF values compared to those in the control group. In contrast to the rats in the L-Thy group, those in the L-Thy + SO2 group exhibited significantly slowed HR and increased LVFS and LVEF values. Besides, the rats in the L-Thy + SO2 + HDX group possessed a significantly faster HR and decreased LVFS and LVEF values in contrast to those in the L-Thy + SO2 group. All these differences above were statistically significant (p < 0.05). In summary, these results demonstrate that high thyroxine leads to changes in the left ventricular structure and impairs the cardiac function in SD rats. However, intervention with SO2 improved the cardiac structure and function of hyperthyroid rats, while this improvement was reversed after HDX intervention.
Effect of SO2 on myocardial fibrosis in hyperthyroid rats
The deposition of myocardial collagen fibers was observed and as displayed in Fig. 3A, B. The results showed that the blue-stained collagen fibers were about 5 times in the L-Thy group compared with those in the control group (p < 0.05). The number of cardiomyocytes was reduced and disordered, and myocardial interstitial collagen deposition was significantly increased. However, in the L-Thy + SO2 group, the area of blue-stained collagen fibers was reduced by about 1/2 compared to that in the L-Thy group (p < 0.05). Besides, the number of cardiomyocytes increased, and they were relatively arranged in order. Compared with the L-Thy + SO2 group, the L-Thy + SO2 + HDX group exhibited a significant increase in the area of blue-stained collagen fibers, approximately 1-fold increase (p < 0.05) and a more disordered arrangement of cardiomyocytes. Meanwhile, changes in expressions of fibrosis-related proteins of Collagen I, Collagen III, MMP2, MMP3, and TIMP2 were evaluated through Western-Blot analysis (Fig. 3C–H). The results showed that the expressions of all these proteins were significantly higher in myocardial tissues in the L-Thy group than those in the control group, while the expression of TIMP2 protein was significantly reduced (p < 0.05). Compared with the L-Thy group, the expressions of Collagen I, Collagen III, MMP2, and MMP3 in the myocardium of rats in the L-Thy + SO2 group were decreased, while the expression of TIMP2 protein was significantly increased (p < 0.05). However, the effect of SO2 on the above protein expressions was significantly reversed in the L-Thy + SO2 + HDX group compared with the L-Thy + SO2 group after HDX intervention (p < 0.05). All these results indicate that SO2 can alleviate the dysregulation of MMPs/TIMPs expression and reduce collagen fiber deposition in myocardial tissues of hyperthyroid rats. Reduced the expression of fibrosis-related proteins in cardiomyocytes. Thus, improving myocardial fibrosis in hyperthyroid rats.
SO2 alleviated the mitochondrial damage and inhibited hyperthyroidism-induced apoptosis in cardiac myocytes of hyperthyroid rats
Fig. 4A–D illustrated the TEM results for the ultrastructure of myocardial tissues of rats in each group. The figures revealed that the myocardial fiber arrangement was disordered in the L-Thy group compared with that in the control group. Meanwhile, the mitochondrial swelling, mitochondrial cristae disappearance, mitochondrial outer membrane rupture, and ER swelling were observed. Compared with the L-Thy group, the myocardial fiber disorder mitochondrial swelling and ER swelling were alleviated in the L-Thy + SO2 group. On the other hand, the results in the L-Thy + SO2 + HDX group were similar to or even more severe than those in the L-Thy group. Apoptosis-related proteins, including BAX, Bcl2, Caspase3, and Caspase9, were detected by Western-Blot, as demonstrated in Fig. 4E–I. The results showed that the expressions of BAX, Caspase3, and Caspase9 were all significantly higher in the L-Thy group compared to those in the control group (p < 0.05), while the Bcl2 was significantly down-regulated (p < 0.05). However, the expressions of these pro-apoptotic proteins were down-regulated and the expression of Bcl2 was up-regulated after SO2 intervention (p < 0.05). In addition, anti-apoptotic effect of SO2 was significantly reversed after intervention with HDX (p < 0.05).
With the in vitro experiments, expressions of BAX and Caspase3 in each group of H9c2 cardiomyocytes were detected using immunofluorescence (Fig. 5A), and the expressions of apoptosis-related proteins were measured by Western-Blot (Fig. 5B–D). The results indicated that BAX was significantly up-regulated in H9c2 cardiomyocytes in the L-Thy group while Bcl2 was significantly down-regulated compared with that in the control group (p < 0.05). Moreover, the immunofluorescence detection revealed a significant increase in the fluorescence intensities of BAX and Caspase3 in this group of cardiomyocytes. However, the BAX was downregulated in the L-Thy + SO2 group after SO2 intervention (p < 0.05), while Bcl2 showed a significant upregulation (p < 0.05). The addition of HDX significantly reversed the above effects of SO2. Similarly, immunofluorescence detection revealed similar changes in expressions of BAX and Caspase3 after SO2 and HDX interventions (p < 0.05). It suggests that thyroxine can induce mitochondrial damage in cardiomyocytic rats and lead to cardiomyocyte apoptosis, while SO2 intervention can alleviate thyroxine-induced mitochondrial damage and antagonize cardiomyocyte apoptosis.
Association of SO2 with ERS-related proteins and its potential role in cardiomyocytes
The changes in expressions of ERS-related proteins in the myocardial tissues of rats in each group were analyzed, as observed by Western-Blot (Fig. 6A–D). The figures disclosed that the expressions of CHOP, GRP78/BIP, and ERP72 were significantly up-regulated in the myocardial tissues of rats in the L-Thy group compared with those in the control group (p < 0.05). Nevertheless, SO2 significantly antagonized the up-regulation of the above proteins (p < 0.05). Meanwhile, the changes in expressions of ERS-related proteins in H9c2 cardiomyocytes were observed by Western-Blot in vitro experiments (Fig. 6E–G). The results revealed that CHOP and ERP72 were significantly upregulated in the L-Thy group compared with those in the control group under high TH stimulation (p < 0.05), but were significantly down-regulated after SO2 intervention (p < 0.05). Furthermore, HDX intervention can reverse the inhibitory effect of SO2 on the above ERS-related proteins in vitro and in vivo experiments (p < 0.05). The results indicate that TH can induce myocardial fibrosis and excessive ERS. Additionally, we observed an association between SO2 and the expression of proteins related to ERS, suggesting a potential protective role of SO2 in cardiomyocytes. However, the exact mediating mechanisms warrant further investigation.
SO2 improved ERS by downregulating MST1, LATS1, and P-YAP to antagonize cardiomyocyte apoptosis
This study observed the changes in expressions of MST1, LATS1, and P-YAP in myocardial tissues of rats in each group, as observed by Western-Blot (Fig. 7A–D). The MST1, LATS1, and P-YAP were significantly up-regulated in the L-Thy group compared with those in the control group (p < 0.05), but were down-regulated in the myocardium of rats in the L-Thy + SO2 group (p < 0.05). Meanwhile, the in vitro experiments by Western-Blot (Fig. 7E–H) indicated that MST1, LATS1, and P-YAP were up-regulated in cardiomyocytes of the L-Thy group compared with those in the control group (p < 0.05), but were down-regulated in L-Thy + SO2 group (p < 0.05). Therefore, the in vitro and in vivo experiments demonstrated that HDX reversed the down-regulation of MST1, LATS1, and P-YAP by SO2 intervention (p < 0.05).
In our in vitro experiments, we further investigated the expression of ERS-related proteins by inhibiting the activation of the Hippo pathway via the transfection of YAP1-specific si-RNA. Our results revealed that (Fig. 7K–L), compared to the control group, the expression of ERP72 protein was significantly upregulated in the L-Thy group (p < 0.05). After transfection with YAP1-specific si-RNA, the expression of ERP72 protein in the L-Thy + si-YAP1-1 group of H9c2 cardiomyocytes was downregulated, similar to the Thy + SO2 group (p < 0.05).
DISCUSSION
Biochemically, hyperthyroidism is a condition characterized by low levels of TSH and high levels of THs (T4 and T3) [32]. THs mainly affect the heart and peripheral vascular system, including increased oxygen consumption of myocardial tissues, vascular resistance, blood volume, cardiac contractility, and HR [8]. Hyperthyroidism can lead to several complications such as HTC and compensated heart failure [33]. HTC is a group of cardiovascular diseases dominated by cardiomyocyte hypertrophy and myocardial fibrosis due to the positive inotropic and chronotropic effects of THs on the heart, as well as elevated adrenergic sensitivity and activation of the renin angiotensin aldosterone system [3]. In this study, a hyperthyroid rat model was established by continuous intraperitoneal injection of L-Thy for 5 weeks. The Masson staining and Western-Blot results revealed that myocardial collagen synthesis was significantly increased in hyperthyroid rats, accompanied by impaired synthesis and clearance of extracellular matrix and obviously increased myocardial fibrosis. Similarly, Bao et al. [29] reported a significant increase in interstitial fibrosis in cardiac myocytes of hyperthyroid rats, which is accompanied by weakened cardiac function and cardiac hypertrophy. In addition, Khamis et al. [34] found significant upregulations of cardiac fibrosis-related genes of SMAD-2, SMAD-3, SMAD-4, TGF-β, and NF-κβ in myocardial tissues of hyperthyroid rats, and severely disturbed fibrotic changes in myocardial histopathology. Myocardial fibrosis is a key part in progression of HTC, and is mainly characterized by increased accumulation of fibrillar collagen in the cardiac interstitium and myocardial structural disorders [35]. Meanwhile, it greatly affects the systolic and diastolic function of the heart and the prognosis of patient [36]. Furthermore, this study showed that myocardial fibrosis is an important pathophysiological change and a potentially important target for intervention in hyperthyroid rats.
Apoptosis, a highly conserved physiological process of programmed cell death, plays an important role in normal organism development and maintenance of overall organismal homeostasis. It is mainly divided into endogenous pathways (mitochondrial pathways), exogenous pathways (death receptor pathways), and ERS-induced apoptotic pathways [15,37]. Cardiomyocytes, which are permanent cells with a limited capacity for regeneration in adults, are particularly susceptible to apoptosis. Apoptosis of cardiomyocytes can lead to the loss of these cells, thus activating myofibroblasts can increase the collagen fiber synthesis, which in turn induces myocardial fibrosis [38,39]. Therefore, effectively inhibiting cardiomyocyte apoptosis is important to prevent and treat myocardial fibrosis. Numerous studies have shown that Caspase3 is one of the most frequently activated cysteases during apoptosis [40,41]. BAX can act as a pro-apoptotic signal to increase cell permeability and promote apoptosis, while Bcl2 is an anti-apoptotic signal to promote cell survival and inhibit apoptosis [42]. In this study, the expressions of Caspase3 and BAX were significantly increased in the myocardial tissues of hyperthyroid rats and H9c2 cardiomyocytes after thyroxine intervention, while the expression of Bcl2 was downregulated. Besides, Wang et al. [7] reported a significant increase in apoptosis in the myocardium of hyperthyroid rats after 4 weeks of intervention with thyroxine T4, indicating that high thyroxine can increase the susceptibility of cardiomyocytes to apoptosis [7]. Teixeira et al. [43] found the Bcl2 was downregulated and the BAX/Bcl2 ratio was increased in the hyperthyroid rat model. Additionally, the hyperthyroidism-induced myocardial fibrosis was associated with increased apoptotic protein signaling [43]. Taken together, these results suggest that high thyroxine can induce apoptosis in cardiomyocytes, leading to myocardial fibrosis in hyperthyroid rats.
ERS refers to the accumulation of large amounts of unfolded or misfolded proteins caused by environmental stress in ER, which may inhibit the synthesis of new proteins [44]. ERS can activate the adaptive unfolded protein response, but prolonged ERS can induce apoptosis [45]. ERS-mediated apoptosis may be triggered by three signaling pathways: the CHOP, Caspase-12, and JNK [46]. This study investigated whether the thyroxine-induced apoptosis is associated with excessive ERS and found that hyper thyroxine intervention can cause ER swelling in myocardial tissues, as observed by TEM. Moreover, CHOP, GRP78/BIP, and ERP72 were significantly upregulated in the myocardial tissues of hyperthyroid rats. CHOP is an important mediator of ERS-induced apoptosis and can be upregulated under severe or sustained ERS. Meanwhile, it can regulate the expressions of many pro- and anti-apoptotic genes [47]. Luo et al. [48] demonstrated experimentally that sustained ERS is associated with cardiomyocyte apoptosis and post-infarction fibrosis. Some other studies have reported that excessive ERS can cause cardiac dilatation and even congestive heart failure and myocardial fibrosis [47,49]. These findings are similar to the observation in the hyperthyroid rat model in this study. The in vitro experiments revealed that hyper thyroxine intervention can result in elevated ERS-related proteins (including CHOP and ERP72) in H9c2 cardiomyocytes. This suggests that high thyroxine can affect the ER homeostasis to cause ERS, which in turn induces apoptosis and myocardial fibrosis in cardiomyocytes. There are few studies on hyperthyroidism-induced ERS in cardiomyocytes. Meng et al. [50] studied the thyroid specimens from five hyperthyroid patients and five healthy people and revealed that the ERS-related proteins are involved in hyperthyroidism. This suggests that thyroxine can cause excessive ERS and may be an important mechanism leading to apoptosis and myocardial fibrosis.
The Hippo pathway plays an essential role in regulating the cell proliferation and apoptosis [51]. It consists of the kinase cascade, MST1/2, LATS1/2, the downstream effectors, and transcriptional co-activators YAP and TAZ [52]. The upregulation of the Hippo pathway can induce apoptosis and reduce cell viability in cardiomyocytes [53], which is closely associated with myocardial fibrosis [54]. This study found that high thyroxine induced apoptosis in cardiomyocytes, significantly increased the levels of MST1, LATS1, and P-YAP in myocardial tissues of hyperthyroid rats as well as MST1 protein in H9c2 cardiomyocytes. These findings suggest that the Hippo pathway may play a crucial role in mediating the thyroxine-induced cardiomyocyte apoptosis. Zhang et al. [55] also demonstrated that MST1 overexpression can enhance the cardiomyocyte apoptosis and aggravate the cardiac dysfunction in diabetic rats. YAP is a key downstream transcription factor of the Hippo signaling pathway and is very important in regulating the cell proliferation and apoptotic signaling. P-YAP can be degraded, while unphosphorylated YAP can enter the nucleus to mediate the regulatory mechanisms that antagonize apoptosis. The findings in this study showed that P-YAP and LATS1 expressions were upregulated in myocardial tissues of hyperthyroid rats and H9c2 cardiomyocytes under high thyroid induction. What’s more, MST1 may promote high thyroxine-induced apoptosis in cardiomyocytes by activating LATS1 and mediating YAP1 phosphorylation. Su et al. [56] reported that in a high glucose environment, MST1 can indirectly mediate the phosphorylation of YAP1 at Ser127 and Ser397 sites through LATS1/2, thus regulating the high glucose-induced apoptosis in cardiomyocytes. These results suggest that excessive ERS can activate the Hippo pathway to induce cardiomyocyte apoptosis and mediate the regulatory mechanisms of myocardial fibrosis in hyperthyroid rats.
Previously, SO2 was regarded as an air pollutant that could endanger human health. However, it has now been recognized as a fourth class of endogenous gas signaling molecules, similar to NO, CO, and H2S [24]. An increasing number of studies have revealed that certain levels of endogenous SO2 can exert myocardial protective effects on various diseases, including hypertension, atherosclerosis, myocardial hypertrophy, diabetic myocardial fibrosis, sepsis-induced cardiac dysfunction, and pulmonary hypertension [57]. AAT is the key enzyme responsible for production of the endogenous SO2 in mammals, which comprises two isozymes: AAT1 and AAT2. AAT1 is predominantly located in the cytoplasm, while AAT2 is mainly found in the mitochondria. Meanwhile, AAT presents the highest activity in cardiac tissues [58]. Numerous studies have demonstrated that endogenous SO2 is involved in regulating cardiac function. For instance, Jin et al. [59] indicated that SO2 donors could inhibit isoprenaline-induced myocardial injury and cardiac dysfunction. According to a previous study, SO2 can inhibit myocardial fibrosis in diabetic rats and is associated with the inhibition of cardiomyocyte apoptosis [28]. Nevertheless, the underlying mechanism by which SO2 antagonizes the myocardial fibrosis remains unclear. This study revealed that expressions of AAT1/2 in myocardial tissue were significantly reduced in hyperthyroid rats, indicating that high thyroxine-induced myocardial fibrosis might be associated with the down-regulation of endogenous SO2. However, administration of exogenous SO2 donors up-regulated the AAT1/2. Zhao et al. [60] also reported a decrease in endogenous SO2 production in cardiomyocytes following high glucose stimulation. The results observed in this study suggested that administration of exogenous SO2 donors significantly improved the myocardial fibrosis in hyperthyroid rats, restricted the ERS-activated, and inhibited apoptosis in cardiomyocytes. These outcomes may be attributed to the antagonistic effect of SO2 on excessive activation of the Hippo pathway. In our in vitro experiments, we confirmed that inhibiting the activation of the Hippo pathway can ameliorate ERS in cardiomyocytes induced by elevated THs. Suggesting that SO2 may improve myocardial fibrosis in hyperthyroid rats by suppressing excessive ERS and Hippo pathway antagonizing cardiomyocyte apoptosis.
In conclusion, this study has demonstrated that SO2 can act as a novel endogenous gas signaling molecule to inhibit excessive ERS and cardiomyocyte apoptosis, thus ameliorating the myocardial fibrosis in hyperthyroid rats. The intrinsic mechanism of this effect may be related to antagonizing excessive activation of the Hippo pathway. Furthermore, this study has suggested that downregulation of endogenous SO2 may be involved in the hyperthyroidism-induced myocardial fibrosis, which can be mediated by the Hippo pathway-mediated apoptosis. In contrast, exogenous SO2 may improve myocardial fibrosis in hyperthyroid rats by inhibiting excessive ERS and antagonizing apoptosis of the overactivated Hippo pathway. These results offer new insights into the mechanism of hyperthyroidism-induced myocardial fibrosis and may aid in the identification of novel therapeutic targets and strategies for HTC. However, the underlying mechanism of endogenous SO2 destabilization that contributes to hyperthyroidism-induced myocardial fibrosis and the strategies for regulating endogenous SO2 homeostasis to inhibit myocardial fibrosis require further investigation.





 XML Download
XML Download