

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gliomas represent the most prevalent primary tumors within the central nervous system (CNS), exhibiting an incidence ranging from 1.9 to 9.6 per 100,000 individuals. This incidence variation is contingent upon factors such as age, sex, ethnicity, and geographic location [1]. Historically, tumor classification has relied upon histopathological characteristics and immunohistochemistry (IHC). However, the clinical outcomes and treatment responses of patients with identical morpho logical tumors often diverge due to disparities in the intrinsic genetic characteristics of the tumors. In 2016, the World Health Organization Classification of Tumors of the Central Nervous System (WHO CNS) introduced an integrated diagnostic approach for gliomas, incorporating both pathological and molecular characteristics [2]. Repeated updates to the classification, known as the 5th edition of the 2021 WHO CNS (WHO CNS5), has substantially revolutionized the glioma classification system. This updated version has led to a fundamental change in the classification environment, primarily through the incorporation of numerous molecular biomarkers [3].
Gliomas have been identified to exhibit several genetic alterations that serve as diagnostic, prognostic, predictive, and potentially therapeutic biomarkers. The most frequently reported genetic variations include mutations in genes such as ATRX, BRAF, CDKN2A/B, IDH, NF1, RB1, TP53, telomerase reverse transcriptase promoter (TERTp), and the methylation status of the MGMT promoter. Furthermore, commonly observed genetic features include copy number alterations such as the codeletion of 1p/19q, EGFR amplification, concurrent gain of chromosome 7 along with loss of chromosome 10 (7+/10-), and rearrangements [3].
Depending on the genetic alterations, numerous molecular experimental methods can be used, including Sanger sequencing, quantitative polymerase chain reaction (qPCR) including methylation-specific polymerase chain reaction (MS-PCR), fluorescence in situ hybridization (FISH), and next generation sequencing (NGS) analysis.
NGS technology was designed to enable sequencing of the whole genome, the whole exome, or specific targeted sequences. However, conducting whole genome or exome sequencing is a costly, time-intensive assay, and especially difficult when using small brain biopsy samples, since significant amounts of DNA are required to perform the assay.
Panel-based targeted NGS is designed to sequence specific genes or genetic regions associated with cancer. This approach enables the detection of various genetic alterations, including single nucleotide variants (SNVs), small insertions/deletions (InDels), copy number variations (CNVs), gene fusions, and intragenic deletions. This methodology allows for a rapid turnaround time and is cost-effective. Panel-based targeted NGS provides insights into potentially actionable genetic aberrations, encompassing integrated molecular diagnosis and mutations (SNVs, InDels), CNVs (including gene amplifications), gene fusions, and intragenic deletions [45]. The purpose of this study is to determine the prevalence of genetic alterations and detect actionable target genes through panel-based targeted NGS analysis in glioma patients.
MATERIALS AND METHODS
Patient samples
We collected and analyzed NGS results from 147 patients who underwent glioma surgery at Seoul St. Mary’s Hospital in South Korea. This study received approval from the Institutional Review Board of Seoul St. Mary’s Hospital (Approval No. KC23RASI0117). The patients were diagnosed with glioma between September 2017 and December 2022. As part of the routine diagnostic procedures prior to NGS analysis for these 147 cases, IHC and molecular pathological studies were conducted. These studies included assessment of IDH1 R132H, ATRX, BRAF V600E, and H3K27M expression through IHC, as well as identification of IDH1, IDH2, and TERTp mutations using Sanger sequencing. Additionally, 1p/19q codeletion was assessed using FISH. Pathological diagnoses were determined based on the WHO CNS 5 criteria, utilizing information from histopathology, IHC, and molecular pathology tests (FISH, Sanger sequencing, and NGS).
Materials
This study was conducted on samples containing tumor cells exceeding 10% as identified through hematoxylin and eosin (H&E) staining slides by pathologists. Formalin-fixed paraffin-embedded (FFPE) tissues underwent microdissection at specific regions indicated by unstained slides. The pipeline for CNV analysis takes into account the percentage of tumor cells, which was calculated using the manufacturer’s genomic segmentation analysis. In the case where the percentage of tumor cells was calculated to be less than 40% in the manufacturer’s analysis, the copy number was calculated by applying the proportion of tumor cells manually entered by the pathologist.
Genomic DNA and RNA isolation and measurement (quantity & quality)
The extraction of genomic nucleic acids was accomplished using the RecoverAll Total Nucleic Acid Isolation Kit (Invitrogen, Waltham, MA, USA; A26069). Quantification and quality assessment of nucleic acids (DNA and RNA) were conducted using the NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA; ND-2000) and the qubit fluorometer (Invitrogen, Q33238). The qubit fluorometer was equipped with the Qubit dsDNA HS test kit (Invitrogen, Q32854) and the Qubit dsRNA HS test kit (Invitrogen, Q32855), as per the manufacturer’s instructions.
Nucleic acid amplification and library preparation
We utilized more than 20 ng of nucleic acid for PCR amplification, following the protocol provided by the manufacturer for the Oncomine Comprehensive Assay (OCA) system version.
During the experimental process, the following reagents were employed for cDNA synthesis: SuperScript VILO cDNA Synthesis Kit in OCA v1 (Invitrogen, 11754050), SuperScript IV VILO Master Mix in OCA v3 (Invitrogen, 11756050), NGS Ion Torrent NGS Reverse Transcription Kit (Ion Torrent, Waltham, MA, USA; A45003), and Uracil-DNA Glycosylase, Heat-Labile (Thermo Scientific, 78310100L) in OCA plus version.
The equipment used for amplification is VeritiPro Thermal Cycler, 96-well (Applied Biosystems, Waltham, MA, USA; A48141). Three types of Oncomine (Waltham, MA, USA) Comprehensive Assay (OCA) panels were used in the experiment according to the manufacturer’s upgraded version. We conducted experiments using OCA v1 (Ion Torrent, A29226) from September 2017 to November 2019, OCA v3 (Ion Torrent, A35805/A35806) from December 2019 to September 2020, and OCA plus version (Ion Torrent, A48577/A48578) from October 2020 to December 2022. The amount and quality of amplified products were identified by the QuantStudio 5 (QS5) Real-Time PCR Instrument (Applied Biosystems, A28569).
Next generation sequencing
We used Ion S5 XL Systems (Ion Torrent) equipment for NGS. This Ion S5 XL Systems include Ion Chef and Ion S5 XL sequencer. This Ion Chef was used for consumables and reagents from Ion 540 Kit-Chef (Ion Torrent, A34541) and Ion 550 Kit-Chef (Ion Torrent, A34541). This Ion S5 XL sequencer was sequenced by loading products on the Ion 540 Chip (Ion Torrent, A27766) and the Ion 550 Chip (Ion Torrent, A34538). The sequencing of this experiment used the Ion 540 Chip (Ion Torrent, A27766) from September 2017 to September 2020, and the Ion 550 Chip (Ion Torrent, A34538) was used from October 2020 to December 2022.
NGS data analysis; oncomine comprehensive assay
OCA v1 encompasses 143 genes, OCA v3 covers 161 genes, and the OCA Plus version incorporates panels targeting more than 500 genes. These panels are designed to identify the prevalence of genetic changes while also reporting clinical pathological features. The panels are designed to detect SNVs, In-Dels, CNVs, gene fusions, and intragenic deletions. The panels were upgraded to NGS OCA v1 in 2017, NGS OCA v3 in December 2019, and NGS OCA plus version in October 2020. It was analyzed using a phased upgrade version of the workflow from Ion Reporter (IR) 5.2 to 5.18. These results analyzed IR 5.2 versions from September 2017 to October 2017, IR 5.4 versions from November 2017 to December 2017, IR 5.6 versions from January 2018 to October 2019, IR 5.10 versions from November 2019 to February 2020, IR 5.12 versions from March 2020 to February 2021, and IR versions from March 2021 to May 2021.
The sequencing raw data were aligned to the hg19 (GRCh37) reference genome. The Torrent Suite (Thermo Fisher Scientific, Waltham, MA, USA) was utilized for initial quality control including chip loading density, median read length, number of mapped reads, on target, mean depth, uniformity and, amplicons reading end-to-end.
The cutoff values of mutations were analyzed to be variant allele frequency (VAF) 5% in NGS OCA v1, v3 and VAF 1.5% in NGS OCA plus version. We read depth >10 and a Phred quality score >20 in order to exclude false positive variants. In order to call the false negative variation of the TERTp gene, it was applied by changing it from the cutoff recommended by the manufacturer. The minimum allele frequency was adjusted to less than 8.6%, and the minimum coverage required on each strand was adjusted to 2 (chr.1295228, chr.1295250). For the identification of CNVs, IR software was employed. A cutoff of MAPD (median of the absolute values of all pairwise differences) <0.5 was applied to assess data usability for CNV analysis. Results were reported when the copy number value exceeded the cutoff of 6 for all CNV detection algorithms (applies to all CNV finding algorithm). And reads of gene fusions to more than 40, but all variations were verified and reported after analysis. Variants were subsequently annotated against 5000 Exomes version 20161108 database, Canonical RefSeq Transcripts v98 in OCA v1/v3, v201 in OCA plus. Oncomine Reporter (Thermo Fisher Scientific, A34298) was used for drug-related information.
Statistical analysis
In our study, two software programs were used to analyze and represent the frequency and correlation of detected variations. The calculation of p-values for correlated genes was performed using Python (version 3.9.12; https://www.python.org/downloads/release/python-3912/), while the Fisher’s exact test executed using the Scipy package (version: 1.7.3; https://pypi.org/project/scipy/1.7.3/). A p-value below 0.05 was deemed to indicate significance between the genes.
RESULT
Patient characteristics
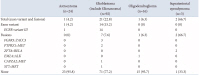
A total of 147 patients diagnosed with gliomas participated in the current study, with 79 (53.7%) identified as male and 68 (46.3%) as female. The median age at the time of glioma diagnosis was 57.4 years, ranging from 4 to 85 years. The majority of gliomas (96.6%) were situated in the cerebrum. Within our cohort, there were 24 (16.4%) low-grade gliomas (CNS WHO grade 1/2) and 123 (83.6%) high-grade gliomas (CNS WHO grade 3/4). A summary of all this information is presented in Table 1.
The classification of all gliomas was performed according to the CNS WHO 5 diagnostic criteria. Specifically, there were 21 astrocytomas, IDH-mutant (CNS WHO grade 2/3/4); 92 glioblastomas (including 1 gliosarcoma) (CNS WHO grade 4); 16 oligodendrogliomas, IDH-mutant and 1p/19q-codeleted (CNS WHO grade 2/3); 3 astrocytomas, IDH-wildtype, NEC (CNS WHO grade 2/3); 2 pilocytic astrocytomas (CNS WHO grade 1); 2 pleomorphic xanthoastrocytomas (CNS WHO grade 2/3); 2 diffuse midline gliomas, H3K27-altered (CNS WHO grade 4); 1 diffuse hemispheric glioma (CNS WHO grade 4); and 8 ependymomas (including 2 ZFTA fusion-positive ependymomas) (CNS WHO grade 2/3), as outlined in Table 2.
Genomic prevalence of somatic variants
A total of 301 mutations (comprising SNVs and InDels) from the glioma cases examined in this study were identified. Specifically, there were 68 mutations in the 24 cases of astrocytomas, 160 mutations in the 92 cases of glioblastomas (including gliosarcoma), 54 mutations in the 16 cases of oligodendrogliomas, 4 mutations in the 2 cases of pilocytic astrocytomas, 2 mutations in the 2 cases of pleomorphic xanthoastrocytomas, 6 mutations in the 2 cases of diffuse midline gliomas, 5 mutations in the 1 case of diffuse hemispheric glioma, and 2 mutations in the 3 cases of supratentorial ependymomas (Table 3).
These 301 mutations were detected in 132 (89.8%) out of the 147 tumors, involving 68 different genes. These genes include ARHGAP35, ARID1A, ARID2, ATRX, ATM, BCOR, BRAF, BRCA2, CDC73, CDKN2A, CIC, CUL4B, CREBBP, DICER1, DNMT3A, DPYD, EGFR, ERCC2, ESR1, FANCA, FANCI, FAT1, FBXW7, FGFR1, FGFR3, FUBP1, H3-3A, H3F3A, IDH1, IDH2, KMT2D, KRAS, MAP2K1, MAP2K2, MAP3K4, MLH1, MSH2, MSH6, NF1, NOTCH1, NOTCH4, NTRK3, PDGFRA, PIK3CA, PIK3CB, PIK3R1, POLD1, POLE, PPP2R1A, PPM1D, PTCH1, PTEN, PTPN11, RAD54L, RB1, SETD2, SMAD4, SMARCA4, SMARCB1, SOX9, STAG2, STK11, TERTp, TET2, TP53, TSC1, TSC2, and ZRSR2. In total, 15 tumor samples (10.2%; 15/147) demonstrated no variants within the targeted genomic regions. Out of the 301 mutations identified across 68 genes, 217 were missense mutations, 37 were frameshift mutations, 42 were nonsense mutations, and 5 affected conserved splice sites.
The 12 genes exhibiting high frequencies were TP53, IDH1, TERTp, PIK3CA, EGFR, NF1, PTEN, ATRX, RB1, BRAF, CIC, and PIK3R1. Mutations were detected in 46 cases (31.3%) for TP53, 35 cases (23.8%) for IDH1, 34 cases (23.1%) for TERTp, 20 cases (13.6%) for PIK3CA, 14 cases (9.5%) for EGFR, 12 cases (8.2%) for NF1, 11 cases (7.5%) for PTEN, 7 cases (4.8%) for ATRX, 6 cases (4.1%) for RB1, and 5 cases (3.4%) for BRAF, CIC, and PIK3R1.
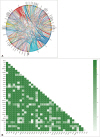
In 24 cases of astrocytoma, 18 types of genetic mutations were detected; IDH1, TP53, TERTp, ATRX, PIK3CA, EGFR, FUBP1, MSH2, CDKN2A, CIC, FBXW7, DNMT3A, NOTCH1, PIK3R1, PTPN11, SETD2, SMARCA4, and NF1. In 92 cases of glioblastoma, 46 types of genetic mutations were detected; TP53, TERTp, PIK3CA, NF1, PTEN, EGFR, RB1, PIK3R1, MSH6, CDKN2A, ERCC2, FANCA, PDGFRA, TSC2, FGFR1, PTPN11, KRAS, PPM1D, POLE, PIK3CB, TSC1, SOX9, PTCH1, STAG2, STK11, SETD2, ARHGAP35, NTRK3, etc. In 31 other cases, 27 types of genetic mutations were detected; IDH1, TERTp, PIK3CA, BRAF, CIC, SMARCA4, NOTCH1, IDH2, H3F3A, etc. The mutation frequency for each diagnosis type is respectively categorized and displayed in Fig. 1.
The TP53 gene was detected in 46 patients, with 3 patients having two TP53 gene mutations each, resulting in a total of 49 detected mutations. Out of the 49 TP53 variants containing multiple gene variants within a single patient case, 44 were missense mutations, 3 were frameshift mutations, and 2 affected splice sites. The most frequent locations for TP53 variants were at codon 248 (8/46, 17.4%), codon 273 (6/46, 13.0%), and codon 175 (3/46, 6.5%). Remarkably, among the 35 IDH1 mutations, 34 (97.1%) involved the p.Arg132His mutation, while 1 (2.9%) involved the p.Arg132Cys mutation. Concerning the 34 TERTp mutations, 26 (76.5%) were associated with the c.-124C>T mutation, and 8 (23.5%) were linked to the c.-146C>T mutation. Regarding the 22 PIK3CA variants that exhibited multiple gene variants within a single patient case, the most frequent locations were codon 344, codon 1043, and codon 1047 (3/22, respectively). Among the 15 EGFR variants with multiple gene variants within a single patient case, they were located at codon 289 (5/15), codon 598 (4/15), and codon 108 (2/15). All 13 NF1 variants were frameshift mutations (8/13) or nonsense mutations (5/13).
Associations of the mutational status between frequently mutated genes
The co-occurrence of mutational states between 68 genes was depicted in Fig. 2A and it shows the combination of 64 genes which mutations are correlated with each other out of a total of 68 genes detected. In these mutated genes, 36 genes significantly correlated mutant states were described in Fig. 2B. IDH1 mutations were showed significant correlation with ATRX, CIC, FUBP1, and NOTCH1 mutant-type (p<0.05). TERTp mutations exhibited correlation with CIC and FUBP1 (p<0.05). PIK3CA mutations demonstrated correlation with FGFR1 (p<0.05).
Genomic prevalence of copy number variations
Out of the 147 tumor samples, evidence of CNVs was observed in 62 samples (42.2%; 62/147) across 25 genes. These genes included EGFR, PDGFRA, CDK4, KIT, MDM4, KDR, MDM2, MET, CDK6, RHEB, PIK3C2B, PIK3CA, AKT3, AR, ARAF, BRAF, CARD11, EZH2, NTRK3, RAC1, RICTOR, RPS6KB1, SMO, SOX2, and KRAS. Conversely, 85 tumor samples (57.8%, 85/147) demonstrated no CNV in the targeted genomic regions.
The 10 genes exhibiting high frequencies were EGFR, PDGFR, CDK4, KIT, MDM4, KDR, MDM2, MET, CDK6, and RHEB. CNVs were only detected in the diagnosis of two types of gliomas: glioblastomas and astrocytomas. One hundred twelve CNVs were identified in 59 (64.1%) of the 92 glioblastomas in 26 genes and in 7 (29.2%) of the 24 astrocytomas in 6 genes. CNVs were identified most frequently in EGFR (18.4%; 27/147 patients), mostly identified at glioblastomas (28.3%; 26/92 patients), followed by PDGFRA (13.6%; 20/147 patients), CDK4 (10.9%; 16/147 patients), and KIT (9.5%; 14 out of 147 patients). Six cases (4.1%) in MDM4, four cases (2.7%) on each of KDR, MET, and MDM2, three cases (2.0%) on each of RHEB and CDK6, two cases (1.4%) on each of PIK3C2B and PIK3CA, one case (0.7%) on each of AKT3, AR, ARAF, BRAF, CARD11, EZH2, NTRK3, RAC1, RICTOR, RPS6KB1, SMO, SOX2, SPOP, and KRAS were detected.
On average, 35 copy number (CN) values were detected across the entire sample, with the distribution ranging from a cutoff of 6 up to 180 (Fig. 3).
Associations of CNV status between altered genes
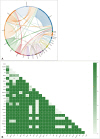
The co-occurrence of the CNV status between 25 genes was demonstrated at Fig. 4A, while the correlation of these 25 genes was shown in Fig. 4B. The CNV status of EGFR showed a significant correlation with CNV status of MDM4 and MDM2 (p<0.05). The CNV status of PDGFRA was correlated with CNV status of CDK4, KIT, and KDR (p<0.05). The CNV status of CDK4 was correlated with CNV status of KIT, MDM2, and PDGFRA (p<0.05). Similarly, the CNV status of KIT was correlated with KDR (p<0.05). The CNV status of MET was correlated with CNV status of BRAF, CARD11, EZH2, SMO, and RAC1 (p<0.05). The CNV status of MDM4 was correlated with EGFR (p<0.05). The CNV status of RHEB was correlated with CNV statuses of AR, ARAF, BRAF, CARD11, EZH2, SMO, and RAC1 (p<0.05). CNV of AR was correlated with ARAF (p<0.05). The CNV statuses of BRAF was correlated with the CNV statuses of MET, CARD11, EZH2, RAC1, and SMO (p<0.05). Similarly, The CNV statuses of EZH2 was correlated with CNV statuses of MET, CARD11, RAC1, and SMO (p<0.05). Finally, the CNV status of RAC1 was correlated with CARD11, and SMO (p<0.05) (Fig. 4B).
Genomic prevalence of gene fusions and intragenic deletions
Twenty-five out of 147 tumor samples (17.0%) showed intragenic deletion in a single gene, EGFRvIII, and gene fusions in six genes: FGFR3::TACC3, PTPRZ1::MET, ZFTA::RELA, EML4::ALK, CAPZA2::MET, and ST7::MET. Conversely, 122 tumor samples (83.0%; 122/147) demonstrated no gene fusions and intragenic deletion in the targeted genomic regions. Intragenic deletion was identified only EGFRvIII (10.2%; 15/147 patients). Gene fusions were detected as follows: FGFR3::TACC3 (2.0%; 3/147 patients), PTPRZ1::MET and ZFTA::RELA (1.4%; 2/147 patients in each gene), and EML4::ALK, CAPZA2::MET, and ST7::MET (0.7%; 1/147 patients in each gene). In this study, gene fusions and intragenic deletion were detected in the diagnosis of 4 types of gliomas: glioblastomas, astrocytomas, supratentorial ependymomas, and oligodendroglioma. Specifically, gene fusions and intragenic deletion were detected in 21 cases (22.8%) of 92 glioblastomas; 14 cases in EGFR-vIII, 3 cases FGFR3::TACC3, 2 cases in PTPRZ1::MET, 1 case on each of CAPZA2::MET and ST7::MET, in 24 astrocytomas; only 1 case (4.2%) in EGFRvIII, in 16 oligodendrogliomas; only 1 case (6.3%) in EML4::ALK, and in 3 supratentorial ependymomas; 2 cases in ZFTA::RELA (66.7%) (Table 4).
Identification of actionable target genes for relevant therapy and clinical trials
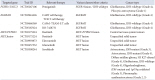
A total of 107 patients (equivalent to 72.8% of the 147 patients) exhibited alterations in the 11 mutated genes (IDH1, BRAF, NF1, EGFR, TERTp, TP53, ATRX, PIK3CA, PIK3R1, PTEN, FGFR), which correspond to FDA-approved therapy, NCCN guideline therapy, and ongoing clinical trials filtering by Oncomine Reporter. Among these, 54 patients (36.7% of the 147 patients) had alterations in the 8 genes (MET, EGFR, PDGFRA, CDK4, KIT, KDR, MDM2, CDK6) associated with CNV for relevant therapeutic considerations. Furthermore, 23 patients (15.6% of the 147 patients) displayed alterations in the 3 genes (FGFR 3::TACC3, EGFRvIII, MET fusion) characterized by gene fusions and intragenic deletion, thus aligning with relevant therapeutic approaches and clinical trial possibilities (as summarized in Tables 5, 6, 7, 8).
DISCUSSION
Historically, the diagnosis of CNS tumors has predominantly relied upon light microscopy. The WHO has formulated criteria for the pathological classification and prognostic stratification of gliomas, primarily grounded in the histological characteristics of CNS tumors. Nevertheless, it is important to recognize that tumors with identical morphological appearances may exhibit distinct molecular profiles, which are closely associated with their biological behavior and clinical outcomes. As a consequence, numerous molecular markers have been incorporated into the assessment of CNS tumor diagnoses. These markers are presently instrumental in guiding patient prognosis and dictating suitable treatment approaches.
As clinically relevant molecular alterations increase, the practicality of single gene analysis is decreasing due to the cost efficiency. Therefore, there is a need for high-throughput techniques that can rapidly evaluate various genetic changes in limited neuropathological samples. Currently, there are several restrictions on the use of exome sequencing in neuropathological samples. A large amount of high-quality DNA is required, but this is limited to obtain from FFPE brain biopsy. It also comes with high costs, long turnaround times. Despite the development of diagnosis and treatment procedures, management of glioma samples is still a difficult task.
As a result, NGS technology has garnered increasing attention and developed rapidly. The advantages of this technology include precise analysis of genetic alterations in tumor samples, cost reduction, and swift diagnosis. Utilizing comprehensive panels that employ FFPE samples in both clinical and research domains, various genetic alterations in gliomas can be simultaneously and rapidly identified. This can contribute to the molecular classification of tumors. Applying the advancements of this new technology allows for the identification of established or potential prognostic biomarkers. These biomarkers can contribute to accurate molecular diagnosis and personalized treatment for glioma patients, thereby enhancing clinical outcomes. Our molecular analysis has demonstrated the potential of NGS targeting gliomas to enhance brain tumor classification and aid clinicians in selecting optimal targeted therapies. However, it is noteworthy that our detection panel had limitations, as it did not encompass the detection of MGMT methylation. This particular aspect needs to be assessed separately using alternative methodologies.
In this study, our findings revealed the presence of 301 mutations (SNVs and InDels) in 89.8% of the tumor samples. Among these mutations, TP53, IDH1, TERTp, PIK3CA, EGFR, NF1, PTEN, ATRX, RB1, BRAF, CIC, and PIK3R1 exhibited high incidences in the mutation spectrum, with TP53 being the most prevalent (31.3%). These results were in alignment with those from other studies [6], as the p53 pathway was found to be deregulated in numerous cases. Moreover, the most frequently detected mutations at codon 248 and 273 of the TP53 gene were observed in TP53-positive tumors at comparable frequencies to those reported elsewhere [7]. Additionally, the p.Arg132His variant was present in 97.1% of IDH1 mutant tumors, consistent with findings reported in other studies [8]. Regarding TERTp mutations (23.1%; 34/147), we observed their presence in oligodendrogliomas (62.5%; 10/16), glioblastomas (20.7%, 19/92), and astrocytomas (20.8%, 5/24). These mutations primarily involved the c.-124C>T and c.-146C>T alterations. TERTp mutations occur in 25% to 70% of gliomas, commonly manifesting at two hotspot locations, c.-124C>T and c.-146C>T [910]. The low detection rate of TERTp mutation is attributed to the poor PCR performance on the GC-rich sequence sites of c.-124C>T and c.-146C>T, resulting in a reduction in the hotspot location detection rate. To compensate for the low detection rate as the importance of TERTp mutations in glioma began to be known, the criteria for calling the hotspot position of the TERTp in the analysis algorithm after analysis from 2020 were modified in this study. The revised content made it possible to analyze lower reads, exceptionally even in severe strand biases, and separately monitor the filtering and exclusion of mutations. In this study, all mutations of EGFR were located in at codon 289, codon 598, and codon 108. Common missense mutations identified in EGFR, which are frequently reported in brain cancer, were clustered in the extracellular domain of proteins and included codon 108, codon 289, and codon 598 [11].
In glioma patients, there are associations of the mutational status between mutated genes. IDH1 is often co-mutated with TP53, ATRX, and NOTCH1 [12]. IDH1 mutations were significantly correlated with ATRX, CIC, FUBP1, and NOTCH1 mutant-type in our study (p<0.05). TERTp mutation, mutations in CIC and FUBP1 have been also known as coexisting mutations for the tumorigenesis of oligodendrogliomas [13]. In our study, TERTp mutations were also significantly correlated with CIC and FUBP1 (p<0.05). PIK3CA mutations were found in 13.6% of our study at the same frequency as other report [14]. In our study, PIK3CA mutations were significantly correlated with FGFR1 in diffuse midline glioma and glioblastoma, IDH-wildtype (p<0.05). PIK3CA are commonly mutated genes in H3F3A-mutant in diffuse midline glioma [15]. Diverse cell signaling pathways are implicated in the initiation and progression of gliomas, including those mediated by EGFR/PI3K/AKT/PTEN, which increase capacity to proliferation, invasion, and cell death [16]. In our study, EGFR mutations were correlated in the PTEN mutation type (p<0.05).
We found that the CNV events were detected only in astrocytomas and glioblastomas. Sixty-two out of 147 tumor samples (42.2%) showed evidence of CNV in 25 genes. CNVs were identified most frequently in EGFR (18.4%) followed by PDGFRA (13.6%), CDK4 (10.9%), KIT (9.5%), and MDM4 (6.5%). The amplification of chromosome 7 with EGFR/MET/CDK6 and chromosome 4 with PDGFRA, are commonly detected in gliomas [17]. The identification of CDK4, KIT, MDM4, PDGFRA, and EGFR amplifications is consistent with previous reports as these genetic changes are frequent in glioblastomas [1819]. However EGFR amplification was less detected in our results (28.3%) comparing prevalence (40%–50%) in previous reports of glioblastomas [20]. The low amplification prevalence of other genes, including EGFR amplification, is due to NGS analysis performed at cutoff 6 or higher of CN value. When the cutoff of the CN value is more than 4, the all CNVs of our study are also detected as 83/147 (56.5%), but we significantly detected the cutoff of ≥6 results compared with the tumor cellularity and other IHC results. The cutoff of CN values 4–6 are also detected in all cases and analyzed comprehensively through the validation of the results of the tumor cellularity, chromosome instability, and IHC staining. Also compared to other regions, lower EGFR amplification rates in Asian patients with glioblastoma were recently reported in screening for ILLANCE1 and ILLANCE2 random glioma trials [21].
Associations of CNV status between altered genes were demonstrated in Fig. 4. In our study, the CNV status of EGFR showed a significant correlation with CNV status of MDM4 (p<0.05). The CNV status of CDK4 was correlated with CNV status of KIT, MDM2, and PDGFRA (p<0.05). The type III subfamily of receptor tyrosine kinases, including PDGFRA, KIT, and KDR (VEGFR), has been reported to play important roles in cell survival, proliferation, and angiogenesis [22]. In our study, CNV of PDGFRA were correlated with CNV of KIT and KDR (p<0.05). Similarly, the CNV status of KIT was correlated with KDR (p<0.05).
Twenty-five out of 147 tumor samples (17.0%) showed evidence of gene fusions and intragenic deletion. Intragenic deletion was identified in EGFRvIII (15/147, 10.2%), as same as previous reports [23]. This EGFRvIII was detected in the diagnosis of two subtypes of astrocytoma and glioblastoma. In this study, gene fusions were identified in FGFR3::TACC3, PTPRZ1::MET, ZFTA::RELA, EML4::ALK, CAPZA2::MET, and ST7::MET (10/147, 6.8%). This gene fusions were detected in the diagnosis of 3 types of gliomas: glioblastomas, oligodendroglioma, and supratentorial ependymomas. FGFR::TACC is best described for FGFR fusion in glioblastoma. In glioblastoma, the FGFR2/3::TACC fusion ratio is detected to be about 2.6%–10%, with most estimates in the 3% range [24]. We found three cases of FGFR::TACC3 fusion (3.2%) in glioblastomas. Frattini et al. [25] described that FGFR3::TACC3 fusion can activate oxidative phosphorylation and mitochondrial biosynthesis and induce sensitivity to inhibitors of oxidative metabolic. This finds oncogenic circuits bound by FGFR::TACC fusion in cancer. The MET fusions were limited to high-grade gliomas. The most common gene fusion was PTPRZ1::MET, followed by ST7::MET and CAPZA2::MET, as same as our results [26]. The PTPRZ1::MET fusion is a recently identified gene fusion of glioblastoma. This fusion occurs as a result of intron insertion and tandem duplication between the PTPRZ1 gene located on chromosome 7q31.32 and the closely located MET gene located on chromosome 7q31.2 [27]. The EML4::ALK fusion has been found in lung, breast, and colorectal cancer. High-grade neuroepithelial tumor with EML4::ALK fusion was also reported by Mrowczynski et al. [28]. We found a case of anaplastic oligodendroglioma with EML4::ALK fusion, suggestive of a novel case. This case had IDH1 mutation, 1p/19q codeletion, TERTp mutation, MGMTp methylation, and PIK-3CA mutations. The histopathology of this case shows oligodendroglioma feature with frequent mitosis (8/10 HPF) and microvascular proliferation. EML4::ALK fusion was analyzed as true positive in the review of raw data. However it cannot be ruled out the possibility of false positive, so additional validation is considered necessary. Recurrent ZFTA::RELA fusions were identified in a large fraction (60%–70%) of supratentorial ependymomas [29]. We found two cases (66.7%) of ZFTA::RELA fusion-positive ependymomas in the three cases of supratentorial ependymoma. RELA fusion-positive ependymoma was newly proposed in the WHO CNS, 2016 updated fourth edition. Fusions of zinc finger translocation associated (ZFTA, formerly known as C11orf95) were shown to not only involve RELA, but also other fusion partners, CTNNA2, MAML2/3, NCOA1/2, or MN1. This tumor was then re-classified as supratentorial ependymoma, ZFTA fusion-positive in the WHO CNS 5 [3].
Recognition of potential actionable targets for patients with glioma is very important. The standard treatment of glioblastomas is based on the Stupp protocol following surgical resection, but typically recurs within 6–9 months of diagnosis. The advantage of NGS testing is that it has potential usefulness in detecting clinically relevant genomic changes in various targetable genes for FDA-approved therapy, NCCN guideline therapy, and clinical trials. In our study, 107 patients (72.8% of 147 patients) had alterations on the 11 targetable mutated genes (IDH1, BRAF, NF1, EGFR, TERTp, FGFR, TP53,
ATRX, PIK3CA, PIK3R1, and PTEN). Fifty-four patients (36.7% of 147 patients) had alterations on the eight targetable genes (MET, EGFR, PDGFRA, CDK4, KIT, KDR, MDM2, and CDK6). Twenty-three patients (15.6% of 147 patients) had alterations on the three targetable genes (FGFR3::TACC3, EGFRvIII, and MET fusion) with gene fusions and intragenic deletion. These results are consistent with previous evidence in various cohorts of glioma patients reporting rates of actionability ranging between 18% and 55% [30]. In our study, we identified several FDA-approved therapy, NCCN guideline therapy, and ongoing clinical trials investigating targeted therapies for actionable genetic alterations in gliomas and malignant solid tumor filtering by Oncomine Reporter (Tables 5, 6, 7, 8). FDA-approved therapy or NCCN guideline therapy for glioma is targeted for IDH1 and BRAF. A few clinical trials investigating targeted therapies for actionable genetic alterations, such as RAF, EGFR, and FGFR in unspecified solid tumor including gliomas are approved in Ministry of Food and Drug Safety of the Republic of Korea [31]. The targeted therapy of vorasidenib for mutant IDH1 demonstrated very encouraging efficacy and minimal toxicity in a recent, randomized phase III trial in patients with low-grade gliomas [3233]. Successful treatment of gliomas with BRAF V600E-mutation with BRAF/MEK inhibitors has been reported as an objective response rate in high-grade gliomas [34]. Therefore, the BRAF V600E test may recur or change clinical management in progressive glioma. No targeted therapies for TERTp mutations in glioblastoma have been approved for treatment. A variety of approaches targeting TERT activity, including small molecule inhibitors, immunotherapy, and vaccines, are under investigation [35]. A clinical trial (NCT05267106) is ongoing to evaluate the efficacy and safety of pemigatinib, a potent and selective FGFR1-3 inhibitor, in participants previously treated for glioblastoma with FGFR1-3 mutation or fusion (Fight-209) [36]. Paxalisib (GDC-0084) is a small molecule with the ability to penetrate the blood–brain barrier (BBB) and inhibit the PI3K/AKT/Mtor pathway, which is associated with improved overall survival [37]. Evidence of antitumour activity was reported in the Phase 1 Ice-CAP trial (NCT03673787) of ipatasertib and atezolizumab in glioblastoma patients with PTEN mutation [38]. EGFR has been the target of numerous clinical trials in glioblastoma patients, but without positive results. Clinical trials are ongoing evaluating regorafenib for recurrent glioblastoma, as well as EGFR or PDGFR inhibitors, such as erlotinib, lapatinib, and nilotinib, or immunotherapy-based methods for EGFR-overexpressing tumors [394041]. Although these drugs show good anticancer effects in many cases, their use in the treatment of CNS tumor is limited. One of reasons for this is the presence of the BBB which affects drug delivery of drugs to the CNS and inter-patient variability [42]. EGFR inhibitors are not so effective in the treatment of EGFR-amplified glioblastoma patients [4344]. Targetable gene fusions and intragenic deletion involving EGFR, FGFR, or MET represent a promising treatment option for malignant glioma. EGFRvIII intragenic deletion in glioblastoma are frequently targeted by immunotherapy. Autologous chimeric antigen receptor (CAR) T cells targeting EGFRvIII (CAR T-EGFRvIII) has been administrated to treat patients with recurrent glioblastoma [4546]. The identification of FGFR3::TACC3 fusion can help identify diffuse glioma patients who are potentially responsive to targeted therapy with FGFR kinase inhibitors [36]. Clinical trial (NCT05267106) to evaluate the efficacy and safety of pemigatinib, a potent and selective FGFR1-3 inhibitor, in participants with previously treated glioblastoma harboring FGFR1-3 mutation or fusion such as FGFR3::TACC3 (Fight-209) is on-going [47]. A randomized controlled open-label multicenter phase II/III clinical trial (NCT03175224) of a MET inhibitor, bozitinib (synonyms; vebreltinib) targeting PTPRZ1::MET fusion is on-going [48]. A Phase I Study (NCT03598244) of savolitinib in recurrent medulloblastoma, high-grade glioma, diffuse midline glioma, and CNS tumors harboring MET amplification or fusion is on-going [49]. Clinical trial (NCT03993873) of elzovantinib in solid tumors with genetic alterations in MET (SHIELD-1) is on-going [50]. Glumetinib, MET inhibitor, in solid tumors is under development to date (NCT03457532) [51].
In conclusion, we present the results of NGS analysis that detected extensive SNVs, small InDels, CNVs, gene fusions, and intragenic deletions in gliomas detailing the frequency and co-occurrence of genetic alterations in patients with gliomas. We also list genetic alterations with potential therapeutic targets and offer a comprehensive discussion of targeted therapy options. The routine use of multigene NGS analysis, which evaluates multiple relevant markers simultaneously, is an effective way to glioma classification and allows for the identification of a greater number of genetic alterations and rare genomic events, leading to more treatment options for glioma patients. Consequently, NGS analysis is required for diagnosis, prognosis, eligibility for clinical trial enrollment, and treatment decisions in patients with glioma.








 XML Download
XML Download