

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Glucose is a crucial fuel for our daily energy needs, and its metabolism must be promptly modulated. The importance of glucose homeostasis is well exempted from type 1 and type 2 diabetes, both resulting from impaired glucose homeostasis and becoming one of the leading aging-associated diseases. Blood glucose levels are tightly controlled by two pancreatic hormones, insulin, and glucagon. Insulin is produced and secreted from pancreatic β-cells. It targets various metabolic tissues, including the brain, immune cells, liver, muscle, and adipose tissue, to modulate carbohydrate, lipid, and amino acid metabolism. In particular, insulin binds to its receptors on the liver, muscle, and adipose tissue to maintain glucose homeostasis. In muscle and adipose tissue, insulin prompts the uptake of surplus blood glucose into both tissues via glucose transporters, whereas insulin suppresses glucose production (gluconeogenesis) in the liver. In contrast to insulin, glucagon is synthesized and released into the bloodstream by pancreatic α-cells. Secreted glucagon binds to its receptor in the liver, promoting hepatic gluconeogenesis.
Insulin deficiency and its associated hyperglycemia are caused by the loss of pancreatic β-cell resulting from either an autoimmune attack on its own β-cell (type 1 diabetes) or insulin resistance-induced β-cell exhaustion (type 2 diabetes). Uncontrolled hyperglycemia leads to other pathophysiologies, such as blindness (diabetic retinopathy), renal disease (diabetic nephropathy), and amputations. In contrast to insulin resistance, which has a variety of therapeutic options, insulin-deficient diabetes imposes a tremendous burden due to its limited therapeutic options.
Since 1949, lithium has been used as a treatment for bipolar disorder [1], and it has also been shown to improve glucose homeostasis by facilitating glucose uptake and glycogen synthesis [2-4]. Glucose transporter 4 (GLUT4, SLC2A4) primarily transports glucose into muscle and adipose tissue, and lithium has been documented to promote GLUT4’s translocation to the plasma membrane of skeletal muscle [2]. Furthermore, lithium and exercise have been demonstrated to reduce body weight and blood glucose levels in obese mice fed a high-fat diet [3]. In type 1 diabetic rodent models that received streptozotocin (STZ) to remove insulin-producing pancreatic β-cells, lithium has been shown to promote the action of ectopically administered insulin to lower blood glucose and has also been proposed to protect β-cell from STZ-induced toxicity [3,5]. However, the metabolic benefit of lithium and its mechanistic understanding in insulin-deficient diabetes, such as type 1 diabetes, remains elusive. Here, we documented that lithium mitigated hyperglycemia in insulin-deficient diabetic mice receiving a high dosage of STZ. Although lithium did not elicit any substantial metabolic changes in the skeletal muscles or any considerable β-cell protection from the toxicity of STZ even after moderate exercise its treatment resulted in a discernible decrease in glucagon-producing α-cell mass. Furthermore, lithium attenuated hepatic gluconeogenic program by decreasing G6pase expression and increasing AMPK activity. In sum, lithium provides metabolic benefits via suppressing glucagon-producing α-cell mass and downregulating hepatic gluconeogenesis in type 1 diabetic conditions, independently of exercise.
METHODS
Animals
C57BL/6J mice (Hyochang Science) were housed under standard conditions with a constant room temperature (22°C ± 1°C), relative humidity (50% ± 10%), and a 12 h light/dark cycle (light from 07:00 to 19:00) with an ad libitum access to food (#2014, Harlan Telkad) and water. All mice were allowed to habituate to the novel environment for 1 week prior to the experiments. All the experimental procedures on the animals were reviewed and approved by the Animal Care and Use Committee of Keimyung University (KM2020-013).
STZ-induced type 1 diabetic mouse
11-week-old male mice were administered intraperitoneally with 400 µl of STZ (50 mg/kg; Sigma-Aldrich, Cat# S0130) or vehicle (0.5M sodium citrate solution; Fisher Scientific, Cat# BP-327-1) after 12-h fasting, which is based on the previous report [6]. 10% sucrose water and normal food were provided immediately to prevent death from hypoglycemia. Mice with over 340 mg/dl of blood glucose, after five-day consecutive injections of STZ, were used for further experimentation with an exercise and/or lithium treatment for additional 12 weeks.
Lithium administration and exercise training
Diabetic mice were introduced with 10 mg/kg of LiCl (Sigma-Aldrich, Cat# L4408) or saline (300 µl) via daily oral gavage one hour before exercise training. The dosage of LiCl was determined based on previous publications [7,8]. Previous studies have shown that 10 mg/kg of Li administration resulted in 0.3–0.7 mmol/L of Li in blood, which is recommended blood level for the treatment of psychiatric diseases [9-11]. We also tested toxicity and observed that long-term Li treatment did not induce marked renal or hepatic toxicity in diet-induced obese rats [3]. LiCl was introduced to mice five days per week for 12 weeks of the experiment. Exercise practice on mice was conducted daily between 09:00 a.m. and 12:00 p.m. for 12 weeks on a treadmill (FT-200; Techman Soft), and its speed and intensity were decided based on the previous reports [12,13] with the following modification: The exercise protocol consisted of low-intensity (40% VO2 max, 17 m/min, slop 0%) walking on a treadmill for 20 min per day, three days per week for 12 weeks. No electric shocks were used to reduce the stress effect of running on the treadmill during training sessions.
Thus, the following five groups were investigated in our study, and detailed experimental conditions were as documented above:
Negative control group (NC): Normal glycemic, saline administered
Positive control group (PC): Hyperglycemic, STZ administered
Lithium-administered group (Li): Lithium administered to STZ-received mice
Exercise training group (Ex): Moderate exercise administered to STZ-received mice
Lithium and exercise group (Li + Ex): Lithium and moderate exercise administered to STZ-received mice
Body weight and food intake were measured daily during the experimental period. After 12 weeks of treatment, mice were rested for 48 h to eliminate the last-bout exercise effect. The mice were fasted overnight, following which they were anesthetized with pentobarbital sodium (5 mg/100 g BW). Blood was collected from the orbital vein using a heparin tube (Micro-Hematocrit capillary tube, #22-362-566; Fisher), and blood glucose was immediately measured using an automatic blood glucose analyzer (YSI 2300; YSI). The remaining blood was centrifuged (1,500 × g, 15 min), and only plasma was stored at −80°C. The plantaris muscles and liver were snap frozen after dissection and stored at −80°C until analysis.
Sample preparation for Western blotting
The plantaris muscles and liver were homogenized together in an ice-cold RIPA buffer (Tris-HCl pH 7.5 50 mM, SDS 0.1%, Triton X-100 1%, sodium chloride 150 mM, sodium deoxycholate 0.5%, EDTA 2 mM; BIOSOLUTION) containing protease and phosphatase inhibitors (#1861280; Fisher Scientific, #P2850; Sigma-Aldrich). Homogenization was performed on ice using a Wheaton tissue grinder (#357535 and #357537; DWK Life Sciences). Homogenates were subjected to three freeze/thaw cycles and centrifuged for 15 min at 4°C and 3,000 rpm in a microcentrifuge (#5425R; Eppendorf). The supernatant was separated and stored at −80°C until assayed.
Western blotting
Protein concentration was determined using the BSA assay (#5000006; Bio-Rad). Aliquots were solubilized in Laemmli buffer (#1610747; Bio-Rad) and subjected to SDS/PAGE. A total of 40 µg of protein was separated by SDS-PAGE gel and transferred to a nitrocellulose membrane. The membrane was blocked for 60 min using 5% non-fat dry milk and Tris-buffered saline with 0.1% Tween 10 (TBST; pH 7.5), washed with TBST, and incubated overnight at 4°C with primary antibodies against the following proteins: calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2, 1:1,000, Cell Signaling, #16810), phospho(Thr180/Tyr182)-p38 mitogen-activated protein kinase (MAPK, 1:1,000, Cell Signaling, #4511), p38 MAPK (1:3,000, Cell Signaling, #9212), GLUT4 (Santa Cruz Bio, #sc-53566), GSK3β (Santa Cruz Bio, #sc-81462), phospho-GSK3β (Santa Cruz Bio, #sc-373800), glycogen synthase (Cell Signaling, #3886), phospho-glycogen synthase (Cell Signaling, #47043), PEPCK (Merck Millipore, #ABC1691), PFK-1 (Santa Cruz Bio, #sc-166722), LDHA (Santa Cruz Bio, #sc-137243), Rab10 (Santa Cruz Bio, #sc-101429), phospho-AKT (Cell Signaling, #4060), AKT (Cell Signaling, #2920), phospho-AMPKα1/2 (Santa Cruz Bio, #sc-33524), AMPKα1/2 (Santa Cruz Bio, #sc-25792), G6Pase (Abcam, #ab93857), and β-actin (Abcam, #ab106814). After washing with TBST, the samples were treated with the secondary antibody (anti-mouse or anti-rabbit, Santa Cruz Biotechnology) for 60 min. The bands were visualized using ECL (Genekhan Scientific), and the relative intensity of the bands was assessed using SigmaGel (Jandel Scientific Corp.).
Immunohistochemistry of the pancreatic islets
The whole frozen pancreas was embedded in FSC22 frozen section media (Leica), and these frozen optimal cutting temperature blocks were sectioned to 6-µm thickness. The sectioned pancreas slices were incubated with insulin (Santa Cruz, #sc8033) and glucagon (Abcam, #ab92517) antibodies at 4°C overnight, followed by washing with PBST (phosphate buffered saline with 0.1% Triton X-100) and subsequently incubating with Alexa-fluor-labeled secondary antibodies (Anti-mouse IgG secondary antibody, Alexa Fluor plus 488 and Anti-rabbit IgG secondary antibody, Alexa Fluor plus 555, Invitrogen) for 3 h at room temperature. Immunofluorescence signals were observed under a fluorescence microscope (Nikon).
We quantified the glucagon-positive area in the islet using CellProfiler (The Broad Institute) [14]. First, we manually selected the islet area from the image, and then we determined the glucagon-positive area from the islet chosen based on the glucagon fluorescence signal.
Statistical analysis
Values are means ± standard error of the mean (S.E.M.). The significance of the differences between groups was assessed using a one-way analysis of variance (ANOVA), followed by a post-hoc comparison using the Tukey significant difference method. A p-value < 0.05 was considered statistically significant. N = 5–8 mice per group were used for analyzing metabolic profiles, and N = 4 mouse samples were used for the quantification of Western blotting. The islet sections of N = 3–4 mice per group were used to quantify glucagon-positive α-cell proportion. Statistical outliners were identified and excluded with GraphPad Prism 9.0 (GraphPad Software).
RESULTS
Exercise or lithium treatment markedly ameliorated STZ-induced hyperglycemia
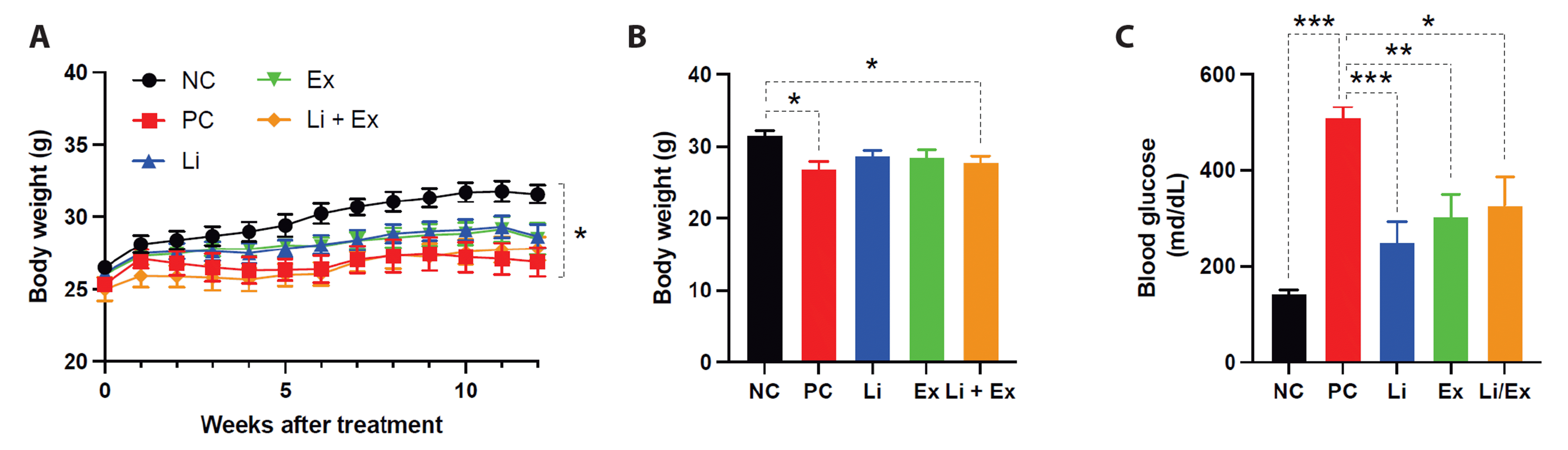
Previously, we have documented that lithium promoted GLUT4’s translocation to the plasma membrane, thereby facilitating glucose uptake in skeletal muscle [2]. Furthermore, lithium facilitated acute exercise-induced glycogen breakdown and glucose uptake in skeletal muscle of mice fed a high-fat diet (type 2 diabetic) and mice with mild type 1 diabetes [3]. This time, we examined whether the combination of lithium and exercise provides metabolic benefits for insulin-deficient type 1 diabetes.
The loss of pancreatic β-cells induced by STZ resulted in body weight reduction and severe hyperglycemia (Fig. 1). Either moderate exercise or lithium administration to type 1 diabetic mice significantly alleviated hyperglycemia without affecting body weight (Fig. 1). However, the co-administration of exercise and lithium did not lead to any synergistic changes in blood glucose levels in comparison to single administration (Fig. 1A, C).
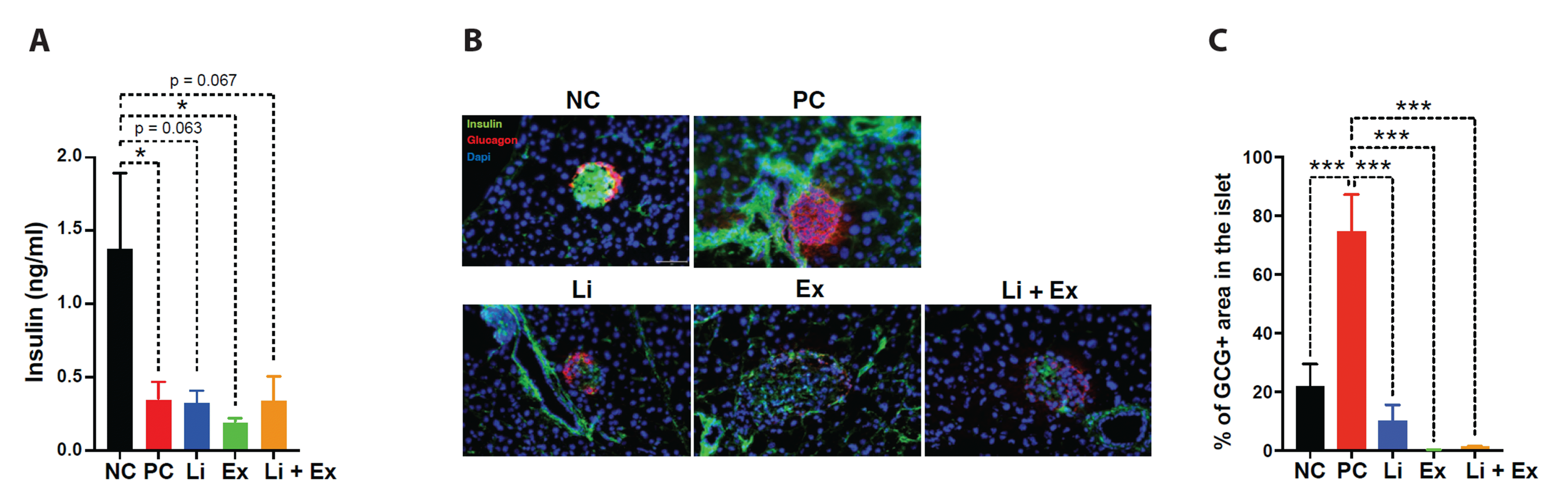
To determine if exercise or lithium protects β-cell loss from STZ toxicity, blood insulin levels and islet histology were examined. STZ caused a decrease in insulin and β-cell mass and an increase in glucagon-producing α-cell mass (Fig. 2). Although neither exercise nor lithium reversed the loss of insulin and β-cell mass induced by high STZ toxicity (Fig. 2A, B), we found that glucagon-positive (GCG+) α-cell mass was significantly reduced after moderate exercise and/or lithium administration (Fig. 2B, C).
Lithium treatment demonstrated limited synergistic metabolic effects of an exercise on skeletal muscle in insulin-deficient type 1 diabetes
Our previous research demonstrated that exercise and/or lithium treatment influenced systematic glucose homeostasis by increasing glucose uptake in skeletal muscle in mice fed a high-fat diet or with mild type 1 diabetes [3]. When we measured the protein levels of factors involved in muscle metabolism, we found that fully insulin-deficient type 1 diabetes had no effect on the protein levels of Glut4 and Rab10 and the phosphorylation levels of Akt and GSK3β (Fig. 3). Lithium did not modulate Glut4 protein levels, despite the fact that exercise significantly increased Glut4 expression (Fig. 3A, B). Intriguingly, co-administration of lithium and exercise eliminated exercise-induced Glut4 expression (Fig. 3A, B). In addition, the protein levels of Rab10, which aids in Glut4 membrane localization, were increased by moderate exercise or lithium treatment (Fig. 3A, B). However, lithium did not augment the effect of an exercise on Rab10 expression in a synergistic manner (Fig. 3A, B). When determining insulin signaling changes after an exercise and/or lithium administration, we observed that phosphorylation of Akt at Ser473 tended to be decreased after STZ treatment, which was reversed after lithium administration, whereas an exercise did not increase Akt phosphorylation in the absence of insulin (Fig. 3A, B). Furthermore, the phosphorylation of GSK3β at Ser9 in STZ-treated mouse muscle was significantly attenuated after exercise, indicating a decrease in glycogen synthesis, which aids in the breakdown of glycogen to provide glucose during an exercise (Fig. 3A, B). Although others have reported that lithium stimulates GSK3β phosphorylation [15], we have not observed an increase in GSK3β phosphorylation following lithium treatment (Fig. 3A, B). However, lithium showed a marked tendency to elevate GSK3β phosphorylation in exercised muscle (Fig. 3A, B).
Following the moderate exercise and/or lithium administration, we investigated additional metabolic signaling and pathways in the skeletal muscle. Although a decrease in glucose uptake (and consequently ATP production) of the skeletal muscle was anticipated after STZ-induced insulin deficiency, we observed only a trend of attenuated AMPK phosphorylation after STZ treatment (Supplementary Fig. 1A, B), which was also reported by others [16,17]. Neither lithium nor exercise altered AMPK phosphorylation (Supplementary Fig. 1A, B). Moreover, CaMKK2 levels, which are known to be involved in AMPK activation and muscle regeneration [18], tended to be lower after STZ treatment (Supplementary Fig. 1A, C). However, CaMKK2 protein levels were unaffected by exercise and/or lithium treatment (Supplementary Fig. 1A, C). Next, we measured the protein expression levels of two key glycolytic enzymes, phosphofructokinase and lactate dehydrogenase A (LDHA), and determined that their expression was not altered by exercise or lithium alone (Supplementary Fig. 1A, D, E). However, the combination of exercise and lithium led to a synergistic increase in LDHA protein levels (Supplementary Fig. 1A, E).
Collectively, in contrast to high-fat diet-fed obese and mild type 1 diabetic conditions, where lithium promoted exercise-induced glycogenolysis and glucose uptake in the skeletal muscles, lithium exhibited no synergistic interaction with exercise in the skeletal muscles under insulin-deficient type 1 diabetic conditions.
Lithium attenuates hepatic gluconeogenesis in insulin-deficient type 1 diabetes independently of exercise training
Since there was a marked reduction of α-cell mass after an exercise and/or lithium treatment, we investigated whether an exercise and/or lithium treatment modulates hepatic gluconeogenesis that is upregulated by α-cell-produced glucagon. In the absence of insulin-producing β-cells as a result of STZ treatment, α-cell mass increased (Fig. 2B), and gluconeogenic enzyme G6Pase protein levels were elevated significantly (Fig. 4A, B). Lithium treatment tended to reduce G6Pase protein levels in the liver of STZ-treated mice (Fig. 4A, B) as its α-cell mass decreased (Fig. 2B). Exercise and the combination of exercise and lithium significantly attenuated liver G6Pase protein levels in mice administered STZ (Fig. 4A, B). PEPCK protein levels did not significantly differ amongst the samples examined (Fig. 4A, C). Next, the regulatory signaling of gluconeogenesis in the liver was evaluated. It is known that AMPK signaling inhibits hepatic gluconeogenesis [19,20]. In the absence of insulin, AMPK phosphorylation at Thr172, which is phosphorylated by AMPK-activating upstream kinases such as LKB1 and CaMKK2 [20-24], markedly decreased (Fig. 4A, D), whereas lithium treatment significantly reversed it (Fig. 4A, D). A moderate amount of exercise had no effect on AMPK phosphorylation, and the combination of exercise and lithium only increased AMPK phosphorylation as much as lithium alone (Fig. 4A, D). In addition, we assessed p38 MAPK activity (phosphorylation at Thr180 and Tyr182) which is known to suppress hepatic glucose production by activating the anti-gluconeogenic endoplasmic reticulum stress signaling component XBP1s [25]. However, we did not notice any changes in p38 MAPK phosphorylation in any of the samples examined (Fig. 4A, D). Furthermore, we did not observe any significant changes in aspartate aminotransferase and alanine aminotransferase activities in the blood, indicating no difference in liver damage after STZ treatment and additional lithium and/or exercise administration (Supplementary Fig. 2).
Collectively, in contrast to obesity and type 2 diabetes, in which lithium facilitated exercise-induced glycogenolysis and glucose uptake in the skeletal muscles, in the absence of insulin, such as in type 1 diabetic conditions, lithium displayed limited synergistic interaction with exercise. Instead, lithium attenuated glucagon-producing α-cell mass and downregulated gluconeogenic programs in the liver, independently of an exercise.
DISCUSSION
Here, we demonstrated that lithium therapy or exercise ameliorated hyperglycemia in STZ-induced type 1 diabetes. However, lithium had no discernible effect on the exercise-induced glucose metabolism of skeletal muscle. This is in striking contrast to previous studies that found lithium to promote exercise-mediated glucose metabolism in skeletal muscle by facilitating glucose uptake and glycogen breakdown in obese and type 2 diabetic mice [2,3]. Given the observed hyperinsulinemia and insulin resistance in obese and type 2 diabetic mice, as opposed to STZ-induced type 1 diabetic mice, the enhancement of glucose metabolism in the muscle in obese and type 2 diabetic mice after lithium administration and exercise may be attributed to the improvement of insulin receptor signaling.
Additionally, we found that lithium and exercise independently decreased glucagon-producing α-cell mass without affecting blood insulin levels and insulin-producing β-cell mass in STZ-treated mice. Next, we examined whether reduced α-cell mass following lithium or exercise administration leads to the downregulation of hepatic gluconeogenesis, hence contributing to glucose-lowering effect of lithium or exercise. We concluded that lithium or exercise independently suppressed hepatic gluconeogenic programs (Fig. 4). Lithium and/or exercise downregulated G6Pase protein levels, one of the key enzymes in gluconeogenesis, although co-administration of both failed to provide a synergic outcome. Lithium was previously documented to attenuate gluconeogenesis directly in in vitro liver cells [26], which also implies that lithium may directly and indirectly (via α-cell) modulate hepatic gluconeogenesis. Interestingly, unlike exercise, which did not increase hepatic AMPK activity via its phosphorylation, lithium significantly reversed the STZ-induced decrease in AMPK phosphorylation. Lithium-induced AMPK phosphorylation was also observed previously in the kidney [27]. Considering AMPK’s established involvement in inhibiting hepatic gluconeogenesis [19,20], our data suggest that lithium, irrespective of exercise, suppresses liver glucose production by raising AMPK activity and also modifying α-cell mass.
On the basis of animal models treated with STZ, it has been reported that α-cell proliferation can be suppressed by γ-aminobutyric acid (GABA) or enhanced incretin action [28,29]. Lithium or physical activity has been documented to augment GABA neurotransmission and its action in the brain [30-33]. In addition, lithium has been shown to increase GLP-1’s action in the brain [34], and physical exercise has been demonstrated to increase GLP-1’s antidiabetic action [35]. It is probable that lithium or exercise influences α-cell mass via GABA or incretin action on the islet.
As the aging and obese populations are growing fast, the incidence of aging-associated diseases, such as type 2 diabetes, has risen dramatically in recent years. However, in contrast to type 1 diabetes, which solely relies on insulin as therapeutics, type 2 diabetes is treatable using a variety of medications. Therefore, developing additional therapeutic options other than insulin is crucial for people with type 1 diabetes. Exercise has been explored as an alternative therapeutic choice for type 1 diabetes and has exhibited a modest but effective improvement in glucose management [36].
Our study discovered lithium’s unique role in glucose regulation in insulin-deficient hyperglycemic conditions, such as type 1 diabetes, by downregulating α-cell mass and lowering glucose synthesis in the liver, both of which are independent of exercise. This implies that lithium could be of significant therapeutic value to people with type 1 diabetes.
 XML Download
XML Download