

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Combined malonic and methylmalonic aciduria (CMAMMA), arising from biallelic variant in the ACSF3 gene that encodes the acyl-CoA synthetase family enzymes, is characterized by elevations of methylmalonic acid (MMA) and malonic acid (MA) in the serum and urine.12 This debilitating condition is highly uncommon; the prevalence rate is approximately 1 in 30,000 individuals.34 The detection of CMAMMA in the general newborn screening test that utilizes a dried blood spot (DBS) to screen for elevated propionyl carnitine (C3) levels is challenging. Therefore, reports of cases of CMAMMA are rare globally, except for those reported in the Neonatal Urine Screening Program conducted in Quebec, Canada.5 We identified CMAMMA in a child who experienced recurrent hospitalizations due to unusually severe and recurrent infections from the neonatal period until 6 months after birth, whom we followed up to 29 months of age. Here, we describe the diagnostic process undertaken to investigate primary immunodeficiency disease (PID) and provide an overview of the patient’s clinical course.
CASE DESCRIPTION
A 52-day-old male infant was admitted to our hospital because of a fever that occurred 3 hours before the emergency department visit. The gestational age was 36 weeks and 5 days, and the birth weight was 2,030 g. He was the 1st baby of dizygotic twins. Apgar scores at 1 and 5 minutes after birth were 9 and 10, respectively. On the 2nd day after birth, the baby experienced tachypnea of 66 breaths/min and was subsequently taken to the emergency room. A cerebrospinal fluid test, included in the routine work-up to exclude central nervous system infection, revealed a possibility of meningitis. Therefore, the baby was hospitalized from the 2nd to the 10th day. However, as no pathogen was found by culture or PCR, the cause of meningitis was not revealed; the baby was discharged without other complications. Vaccinations against tuberculosis and hepatitis B virus were administered according to routine recommendations in South Korea.
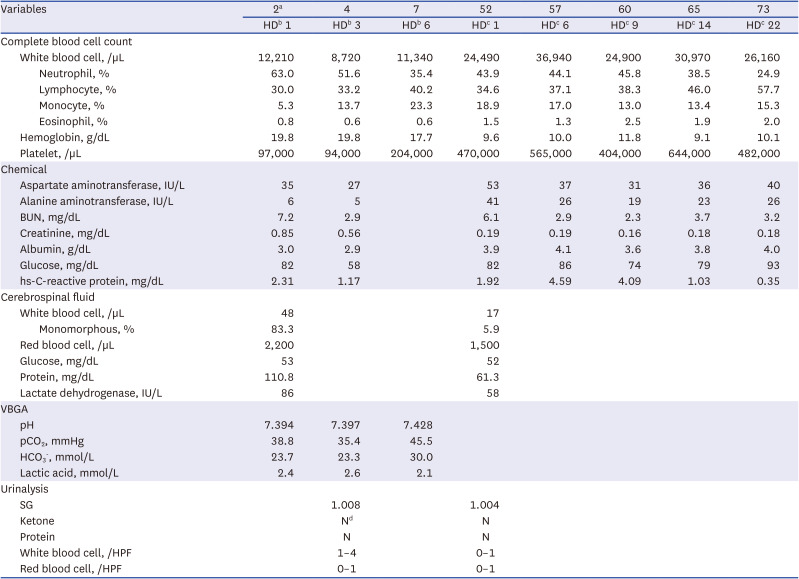
At the current visit, 52 days old, the caregiver reported no complaints except fever. The infant, who was born as a premature twin with anthropometric values below the 0.1st percentile, came to the hospital with a good catch-up growth and showed a height of 51 cm (3rd percentile), weight of 4.4 kg (45th percentile) and head circumference of 37.8 cm (67th percentile) without microcephaly. Upon examination, the infant appeared healthy and active. Initial vital signs were as follows: heart rate, 194 beats/min; respiration rate, 52 breaths/min; body temperature, 38.1°C; and pulse oximetry saturation, 100% in room air. No signs of fontanelle bulging were observed. Thorough physical examinations, including those of the umbilical area, were performed. However, no focal signs indicated current fever. Thorough laboratory work-ups were performed per the infant's medical history and age (Table 1). As the possibility of bacterial meningitis could not be ruled out, empirical antibiotics, including meningitis-dose vancomycin and cefotaxime, were initiated.
Table 1
Laboratory findings in a patient diagnosed of combined malonic and methylmalonic aciduria
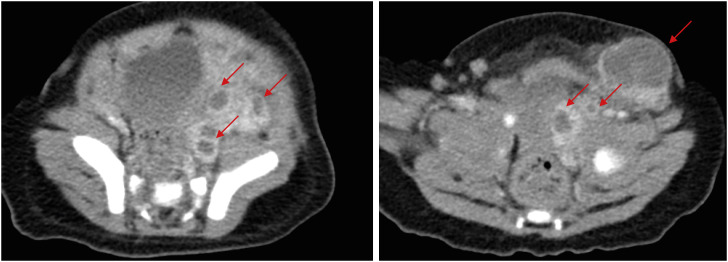
Although the fever at admission persisted at a frequency of 2–3 times a day with a maximum temperature of 38.1–38.5°C, no specific symptoms were observed until hospital day (HD) 5. On HD 5, a 1.5–2 cm-sized mass-like lesion was noted in the left inguinal area. The mass was moderately firm, with signs of inflammation, including redness, swelling, and tenderness. On HD 7, computed tomography (CT) was performed due to a soft-tissue mass-like lesion in the left pelvic cavity, suggesting necrotizing lymphadenitis (Fig. 1). Owing to the possibility of an intra-abdominal abscess with an uncertain pathogen origin, the antibiotics were changed to cefazolin and piperacillin/tazobactam. Because the lesion visualized on the CT was suspected to be a collection of abscesses, incision and drainage (I&D) was performed by a general surgeon. The lesions were dressed daily. On HD 9, as the microbiological culture results revealed the growth of methicillin-resistant Staphylococcus aureus, the antibiotic was changed from cefazolin to vancomycin. On HD 10, the patient experienced defervescence. The patient was discharged on HD 26 after the continuous administration of vancomycin and several follow-up images. One month after discharge, no specific lesions were noted in the inguinal area, and the patient was in good condition.
Fig. 1
Computed tomography images of multiple intra-abdominal abscesses (arrows) in an infant diagnosed with combined malonic and methylmalonic aciduria. The size of the largest abscess in the left inguinal area was 26.10 mm.
The patient was readmitted at 6 months of age due to mild fever and a 1–2 cm-sized round mass with redness and inflammation in the perianal area. Computed work-up by CT revealed abscess formation in the perianal area. Although another I&D was scheduled, the abscess ruptured spontaneously. The infant was given a sitz bath, as recommended by the general surgeon, even though he experienced difficulty. Based on previous findings, vancomycin with piperacillin/tazobactam was initially administered. However, as methicillin-sensitive S. aureus and Escherichia coli were cultured, and the anal lesion showed marked improvement, the antibiotics were discontinued. The patient was discharged on HD 4 without any complications.
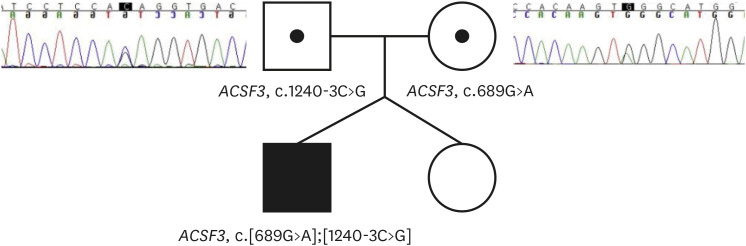
Several unusual bacterial and nonbacterial infections have led us to suspect the possibility of PID. The following tests were conducted: dihydrorhodamine test, immunoglobulin G/A/M, T Lymphocyte cell subset, and next-generation sequencing (NGS) gene panel for PID (NGS-PID panel, GC Labs, Yongin, Korea). All tests except for the NGS panel showed negative results. The NGS–PID panel identified a pathogenic variant (PV) and a variant of uncertain significance (VUS) both in the ACSF3 gene (NM_174917.5), which encodes acyl-CoA synthetases. The PV in the ACSF3 gene indicated CMAMMA, as retrieved from the Online Mendelian Inheritance in Man. The patient's parents were genetically evaluated as CMAMMA is inherited in an autosomal recessive manner. The PV (c.689G>A(p.Trp230Ter)) was detected in the mother, whereas the VUS (c.1240-3C>G(p.?)) was detected in the patient’s father (Fig. 2). Serum amino acid and acylcarnitine testing revealed no significant findings. Still, urine organic acid analysis demonstrated elevated levels of MA at 49.2 mmol/mol (reference range, 0 mmol/mol of creatinine) and MMA levels at 332.1 mmol/mol (reference range, < 32.9 mmol/mol of creatinine), resulting in an MMA/MA ratio of 6.75.
Fig. 2
Patient’s family pedigree and Sanger sequencing results of the patient’s parents. The patient was found to have inherited a compound heterozygous type of mutation, receiving different mutations from each parent.
PV and VUS were detected in each allele of this patient, but experimental confirmation in vitro or in animals could not be performed to clarify the meaning of VUS. However, since clinical specimens showed increased malonic and MMAs, the observed VUS could be considered a novel genetic test result with a high possibility of PV. Thus, we concluded that the compound heterozygous variants of ACSF3 were responsible for CMAMMA in the patient.
The patient has been followed-up until recently, at the age of 29 months. The patient showed normal growth and development without any specific dietary modifications. The patient attends daycare centers and occasionally catches a cold. However, to date, no severe bacterial infections have been reported.
DISCUSSION
CMAMMA is caused by homozygous or compound heterozygous variants in the ACSF3 gene and is a rare disease with a prevalence of approximately 1 in 30,000. Approximately 50 cases have been documented worldwide, highlighting its rarity in the global population, although its actual prevalence remains unknown. The ACSF3 gene encodes a putative methylmalonyl-CoA and malonyl-CoA synthetase, which belongs to the acyl-CoA synthetase family.1 This enzyme is responsible for converting MA and MMA into their respective CoA forms. Therefore, the deficiency of this enzyme leads to the accumulation of MA and MMA in the body. One of the distinguishing features of CMAMMA is the increased ratio of MMA to MA compared with that in individuals with normal malonyl-CoA decarboxylase activity. However, patients with CMAMMA have normal levels of C3, making it challenging to detect this condition in screening tests unless MMA and MA are specifically chosen as primary screening markers. The most comprehensive report, which documents a cohort from Quebec, Canada, conducting newborn urine screening tests, includes 25 patients diagnosed with CMAMMA.6 The characteristic of CMAMMA not being diagnosed in routine newborn screening tests utilizing DBS suggests a significant possibility of underdiagnosis among patients. Therefore, it is highly probable that a considerable number of patients will never be diagnosed for their entire life globally, not only in Korea.
The symptoms of CMAMMA vary widely across affected individuals. In children, symptoms commonly include developmental delays, failure to thrive, and potential complications such as ketoacidosis, which can damage tissues and organs because of excessively acidic blood.5 Other frequently observed symptoms in children include dystonia, hypotonia, developmental delay, hypoglycemia, and microcephaly. However, some patients with CMAMMA may remain asymptomatic until adulthood. In these cases, psychiatric features and neurological problems resembling Alzheimer’s disease and multiple sclerosis may arise, along with seizures, memory loss, cognitive decline, and psychiatric disorders.57 Owing to the wide spectrum of symptoms and variable age of onset, which could range from infancy to adulthood, it is extremely challenging to suspect and proceed with testing for metabolic disorders based solely on symptoms. Considering our patient’s characteristic history of multiple infections since infancy, additional genetic testing, such as primary immunodeficiency disease NGS or whole-exome/genome sequencing, was required. Without such supplementary genetic tests, a diagnosis of CMAMMA would have been impossible.
Determining whether severe infection in CMAMMA is caused by vulnerability to the infection itself or post-infection deterioration of the metabolic state, such as metabolic acidosis, is challenging. However, when compared with MMA, which has similar metabolite and clinical features to CMAMMA, it is thought that both mechanisms may be involved. Much of the reason MMA is susceptible to infection is that elevated MMA reduces mitochondrial function that regulates immune pathways during infection. Therefore, patients with MMA also have similar clinical features to that of primary mitochondrial disorders (hypoglycemia, metabolic acidosis, developmental delay, failure to thrive, hypotonia, microcephaly). Like MMA, CMAMMA also exhibits a wide range of clinical symptoms similar to mitochondrial diseases, including infection. Therefore, it is speculated that CMAMMA is also vulnerable to infection due to the degradation of mitochondrial function due to the accumulation of MA and MMA and the deterioration of various immune mechanisms induced from there, but further experimental data must be accumulated enough for accurate determination. In contrast, the leukocytosis observed when our patient was infected was a completely different clinical picture from the myelosuppression commonly seen in MMA patients when infected. As the infection status improved, leukocytosis gradually improved and reached a near-normal value. Regardless, this difference also suggests that the mechanisms that cause vulnerability to infection may be different between the two diseases.
Currently, there is no definitive treatment for CMAMMA other than dietary management, which involves restricting protein intake and increasing carbohydrate consumption.2 No specific treatment has been proven to have a clear efficacy for managing CMAMMA. Our patient had continuously been administered L-carnitine and mecobalamin since CMAMMA was diagnosed at nine months. It is uncertain whether medication intake improved the patient’s clinical presentation. Serum amino acid and serum acylcarnitine tests showed no significant abnormalities at the time of diagnosis or the 29-month follow-up. In contrast, urine organic acid analysis revealed a decrease in MA from 49.2 to 25.8 and an increase in MMA from 332.1 to 407.7, leading to a significant increase in the MMA/MA ratio from 6.75 to 15.8. Although the exact meaning of the MMA/MA ratio remains unclear, it can be regarded as a potential tool for evaluating the treatment response in CMAMMA.2 Specifically, if an increase in the MMA/MA ratio correlates with a decrease in the clearance of MMA, it can indicate treatment efficacy. Therefore, based on the urine organic analysis results, whether the medication improved the symptoms is unclear. In addition, it is difficult to interpret the MMA/MA ratio increase and judge the effectiveness of L-carnitine because they did not eat the recommended protein-restricted diet, and there are differences in individual diet composition in each family. However, considering the possibility that the patient had not experienced any crisis and had shown good growth during this period, the potential efficacy of L-carnitine and mecobalamin cannot be completely ruled out.
CMAMMA is rarely reported in Asians. The only study from Asia was conducted in China, including the largest number of Asian patients (three individuals in 2021).8 It is unclear whether the disease is more prevalent in Western countries. However, considering the benign course of the disease in most affected individuals, a large population of people with the disease remains undiagnosed. Interestingly, there were similarities between the variants observed in our patients and those reported in a Chinese study. The c.689G>A missense mutation identified in our patient was the same as that reported in Chinese patients. In contrast, the c.1240-3C>G missense mutation was a novel variant that has not been previously reported. A total of four patients have been diagnosed in East Asia so far. As more data are accumulated later, racially specific data is expected to be analyzed, like the paper published in Canada.
By analyzing the clinical manifestations of patients reported thus far, it is uncommon for cases to progress to a metabolic crisis accompanied by metabolic acidosis. Moreover, these symptoms often resolve spontaneously after infancy. Our patient also demonstrated normal growth and development, and the absence of severe infections after six months of age suggests that he will likely follow a course similar to that of previously reported cases. However, the possibility of neurological symptoms emerging during adulthood needs to be considered, and long-term follow-up observations must be conducted.910
In hindsight, without the suspicion of other diseases, including PID, the patient would not have been diagnosed with CMAMMA for the remainder of his life. Clinical suspicions by the attending physician and progress in molecular genomics have led to the diagnosis of a rarely reported disease because of the inherent challenges of the disease itself. Through this case, it was realized that the usefulness of urine organic acid analysis is great in confirming the presence of underlying metabolic diseases, including CMAMMA, in patients with suspected PID. Suppose the patient is suspected of having PID due to recurrent or serious infection in the infant period or shows clinical features similar to mitochondrial disease that invades multiple organs. In that case, it is cheaper than genetic testing. It is not invasive and can confirm results quickly. If urine organic acid analysis is combined with genetic testing, we think it will help make a diagnosis quickly and accurately.
 XML Download
XML Download