

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diffuse intrinsic pontine glioma (DIPG), now commonly known as diffuse midline glioma (DMG), is a type of glioma that originates from glial cells, the supportive cells that surround and protect neurons in the brain [1]. DMG/DIPG is a highly aggressive and lethal pediatric cancer that primarily affects children between the ages of 6 and 9 regardless of gender [23]. This type of tumor is characterized by its location in the pons, a part of the brainstem responsible for critical functions such as breathing, heartbeat, and consciousness. It is diagnosed in approximately 150–400 children each year in the United States [3]. The most common symptoms of DMG/DIPG are coordination difficulties, weakness in the limbs, trouble speaking, and vision problems. These symptoms arise due to the tumor’s proximity to vital areas of the brain, which prevents surgical removal of the tumor [4]. The current standard of care for DMG/DIPG is radiation therapy, which can provide temporary relief of symptoms, but does not improve survival rates [5]. Because of the aggressive nature of this disease, the survival rate of children with DMG/DIPG is low, and most patients succumb to the disease within one year of diagnosis [67]. Research on DMG/DIPG is limited by the rarity of the disease and the difficulty in obtaining tissue samples. However, recent advances in molecular and genetic analyses have improved our understanding of the underlying biology of DMG/DIPG. Studies found that DMG/DIPG tumors contain a specific K27M mutation in histone H3 (H3K27M). Because the mutation leads to a distinct and relatively uniform phenotype with significant genetic, epigenetic, and clinical implications, recent research and ongoing clinical trials are focused on histone mutation and epigenetic changes. This review provides a brief overview of the epigenetic and metabolic changes associated with H3K27M mutations. Additionally, we summarize the promising therapeutic targets currently under investigation for the treatment of DMG/DIPG.
DMG/DIPG AND EPIGENETICS
Although the exact cause of DMG/DIPG is not clear, recent research identified a specific genetic mutation on histone H3 that is present in a significant proportion of DMG/DIPG cases [8910]. Histones are involved in the packaging of DNA in eukaryotic cells. DNA is wrapped around histone proteins to form a complex called a nucleosome, which is the basic unit of chromatin and is composed of an octamer of histone proteins (two copies each of histones H2A, H2B, H3, and H4) [11]. In 80% of DMG/DIPG tumors, lysine 27 in either histone H3C2 (H3.1) or H3-3A (H3.3) is mutated to methionine [891213]. Specifically, the mutation occurs in genes that code for histone H3 proteins, which are critical for regulating gene expression and maintaining the stability of DNA within cells. The mutation causes a change in the structure of the histone protein, which alters its ability to interact with DNA and other proteins. This, in turn, can lead to changes in gene expression that promote the growth and spread of cancer cells [14]. In order to acknowledge alternative mechanisms that can modify the pathogenic pathway in DMG/DIPG, this tumor was reclassified as “H3 K27-altered” instead of “H3 K27M-mutant” in 2021 WHO Classification of Tumors of the Central Nervous System [1516].
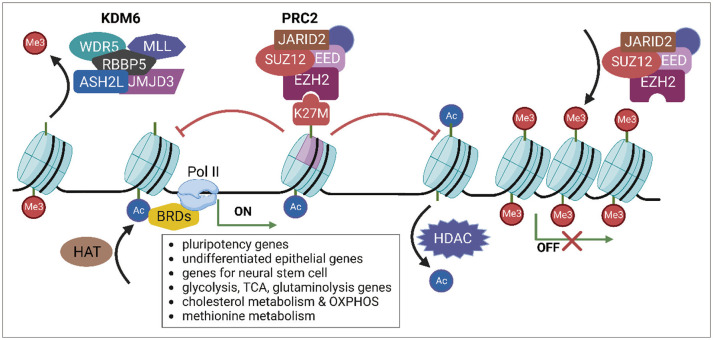
Due to the presence of multiple variant forms of H3, DMG/DIPG cells with H3K27 mutations have a mixture of wild-type and mutant forms of the H3 protein. Although the H3K27M mutation retains only 3%–17% of the entire histone H3 protein, it can lead to important epigenetic changes in DMG/DIPG [17]. One of the epigenetic changes caused by the histone H3 gene mutation is a decrease in the level of H3K27me3, which is a specific type of histone modification that plays a vital role in regulating gene expression by post-translational modification (PTM) (Fig. 1) [171819]. It is involved in a process called epigenetic regulation, which controls how genes are turned on or off without changing the underlying DNA sequence. The diminished methylation level of H3K27 is essential for glioma tumorigenesis [18]. Altered H3K27me3 levels in DMG/DIPG tumors may contribute to the development and progression of this aggressive cancer because H3K27me3 represses the expression of genes associated with cancer development, such as those involved in cell proliferation and survival [20]. The H3K27M mutation promotes the self-renewal of neural stem cells and is associated with neurodevelopmental changes in DMG/DIPG [212223]. In addition, markers of undifferentiated human embryonic stem cells (POU5F1, SOX2) and undifferentiated epithelium (E-cadherin) are maintained at elevated levels in cells with the H3K27M mutation (Fig. 1) [2425].
The H3K27M mutation affects the function of polycomb repressive complex 2 (PRC2) [26]. In normal cells, PRC2 regulates gene expression by adding the methyl group to the H3 protein at specific locations, including H3K27. This modification is associated with a repressed chromatin state, which inhibits the expression of genes in that region of the genome [27]. However, in cells with the H3K27M mutation, PRC2 strongly binds to this mutation and cannot properly add the methyl group to the nascent H3 protein at H3K27 (Fig. 1). In addition, isoleucine as well as methionine strongly binds to and inhibits PRC2 [17]. As a result, the affected chromatin regions become constitutively active, leading to the dysregulation of gene expression and ultimately promoting the development and progression of cancer. However, not all genes regulated by H3K27me3 are decreased in the presence of the H3K27M mutation. The K27M mutation causes a non-random decrease in the deposition of H3K27me3, and only residual levels are present. The remaining H3K27me3 deposition is confined to large unmethylated CpG islands that are enriched for PRC2. Removal of the K27M mutation using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 allows PRC2 to deposit H3K27me3 in sites that have lost it, such as intergenic sites and partially methylated domains with poor PRC2 recruitment. This uninhibited PRC2 activity restores the deposition of H3K27me3 from the large CpG islands [1828].
The H3K27M mutation causes an increase in the level of H3K27ac, a PTM related to active transcription, concomitant with the decrease in H3K27me3. Heterotypic H3K27M-K27ac colocalizes with the bromodomain proteins at transcriptionally activated genes. Bromodomain-containing proteins (BRDs) contribute to activating transcription by recruiting RNA polymerase II (RNA pol II) (Fig. 1) [2930]. Similar to H3K27me3, not all genes that are marked with H3K27ac are affected by the presence of the H3K27M mutation. In DMG/DIPG cells in which H3.3K27M is knocked out using CRISPR-Cas9, most H3K27ac sites (96.6%) remain unchanged compared with the parental isogenic clones [31].
Controversial findings regarding changes in other epigenetic markers in the presence of the H3K27M mutation have been reported. One study found that silencing the H3K27M mutation in DMG/DIPG xenografts restores H3K27me3 levels that were lost due to the mutation, whereas the levels of H3K4me3 and H3K36me3 remain unaffected [32]. However, another study that used single-molecule imaging and genome-wide profiling techniques to analyze combinatorial chromatin patterns demonstrated that the H3K27M mutation has a direct impact on the MLL1-H3K4me3 pathway, leading to a widespread redistribution of H3K4me3 throughout the genome [33]. Mass spectrometry analysis of DMG/DIPG cell lines with CRISPR-edited H3K27M-knockout showed a consistent reduction in H3K9me3 levels, as well as a pattern of H3K36me3 enrichment across the gene bodies of highly expressed genes [28].
The existence of epigenetic heterogeneity in DMG/DIPG was recently proposed. Even within a single cell line, two distinct epigenetic subpopulations were identified in H3K27M-mutated cells. These subpopulations were consistently observed across tumor cell lines derived from different patients and showed differential expression of stem cell and differentiation markers, as well as differences in proliferation capacity. It suggests a potential threshold effect for mediating various epigenetic alterations. The study also demonstrated that different H3K27M levels have different effects on various histone modifications, including H3K9me2, H3K9me3, H3K36me2, and cleaved H3 in individual cells [2034].
Histone markers are important regulators of gene expression because they control the accessibility of DNA to the transcriptional machinery. Specific modifications, such as H3K4me3 and H3K36me3, are associated with active gene transcription, whereas others, such as H3K27me3 and H3K9me3, are associated with gene silencing [35]. Dysregulation of these histone markers can silence tumor suppressor genes or activate oncogenes, leading to uncontrolled cell proliferation and tumor growth [14]. Dysregulation of histone markers also affects genes involved in metabolism in several types of cancer. These metabolic genes are involved in processes such as glycolysis, oxidative phosphorylation, and fatty acid synthesis, and their dysregulation can lead to altered energy metabolism in cancer cells, which is a hallmark of cancer.
DMG/DIPG AND METABOLISM
Metabolic alterations in DMG/DIPG cells promote their growth and survival, a characteristic of cancer. One critical metabolic alteration in DMG/DIPG cells is increased reliance on glucose for energy. Glucose transporters and enzymes involved in glycolysis, which is the breakdown of glucose into pyruvate, are upregulated in DMG/DIPG, suggesting that DMG/DIPG cells have a high demand for glucose to fuel growth and survival. A specific genetic mutation in DMG/DIPG cells is associated with this glucose dependence. The glucose dependence of DMG/DIPG was confirmed in several studies. For example, a study published in Neuro-Oncology Advance in 2020 found that blocking glucose uptake in DMG/DIPG cells slows tumor growth and improves survival in animal models [36]. However, other metabolic alterations in DMG/DIPG cells contribute to their aggressive behavior. For example, DMG/DIPG cells have increased levels of lactate dehydrogenase, which converts pyruvate into lactate. This suggests that DMG/DIPG cells have an increased reliance on anaerobic glycolysis, which can provide energy in the absence of oxygen [37].
Other metabolic alternations in DMG/DIPG cells include increased cholesterol biosynthesis, which begins with acetyl-CoA, and mitochondrial oxidative phosphorylation, which is dependent on the adenosine triphosphate (ATP)-generating pathway [38]. DMG/DIPG cells also show an increase in the tricarboxylic acid cycle metabolism as well as the upregulation of several metabolites such as alpha-ketoglutarate, which is produced by isocitrate dehydrogenase and maintains a preferred epigenetic state of low H3K27me3 [39].
DMG/DIPG cells exhibit increased uptake of glutamine, an amino acid involved in several physiological processes in the body including the synthesis of proteins, neurotransmitters, and nucleotides. Glutamine metabolism may play a role in the development and progression of DMG/DIPG, and targeting glutamine metabolism has been proposed as a potential therapeutic strategy for this disease [40]. One approach is the use of drugs that inhibit enzymes involved in glutamine metabolism, such as glutaminase [39]. These drugs have shown promising results in preclinical studies, reducing the growth and viability of DMG/DIPG cells. Another approach is restricting the availability of glutamine in DMG/DIPG cells, which can be achieved by feeding a low-glutamine diet or by pharmacologically interfering with glutamine uptake [39].
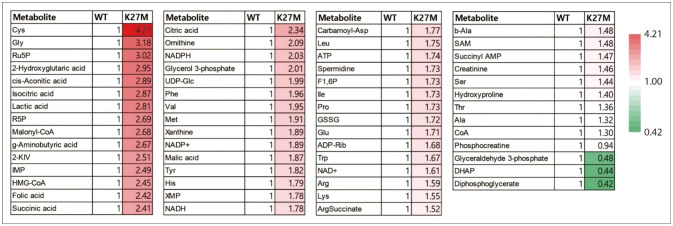
Other metabolic changes in DMG/DIPG have been reported. Because DMG/DIPG cells show a high dependence on methionine, silencing methionine adenosyltransferase 2A (MAT2A) or restricting methionine availability is a potential therapeutic strategy, leading to a change in the epigenome and transcriptome [41]. The metabolic changes in DMG/DIPG were summarized in Table 1. Also, we analyzed published data from Neuro-Oncology Advances and visualized the alteration of metabolites in murine glial cells expressing H3K27M (Fig. 2) [36]. Overall, these findings suggest that targeting specific metabolic pathways in DMG/DIPG cells may be a promising strategy for the treatment of this aggressive tumor.
TREATMENT BY EPIGENETIC OR METABOLOMIC REGULATION
Despite active research efforts, there is currently no cure for DMG/DIPG, and treatment options are limited. The standard of care for DMG/DIPG includes radiation therapy, which is typically used to manage symptoms such as pain and difficulty with motor function. Radiation therapy can reduce the size of the tumor and alleviate some of the symptoms, but it is not a cure [542]. Ongoing clinical trials are exploring new treatments for DMG/DIPG, including targeted therapies, immunotherapy, and gene therapy [542434445]. Some of these treatments target specific molecular pathways involved in the growth and survival of DMG/DIPG cells. Genetic and molecular approaches are essential to identify new and effective therapeutic strategies for this devastating disease.
Targeting H3K27me3 and the pathways it regulates is an area of active research in DMG/DIPG. One approach is the design of drugs that specifically inhibit PRC2 or other enzymes involved in histone modification. Because PRC2 is essential for the proliferation of tumors expressing H3K27M, targeting PRC2 could be a promising therapeutic approach for the treatment of these tumors. Targeting these enzymes may restore normal H3K27me3 levels and repress the expression of genes that promote tumor growth. Inhibition of EZH2, a catalytic subunit of PRC2, affects the proliferation of patient-derived DMG/DIPG cell lines by inducing CDKN2A expression. The strong dependency of DMG/DIPG on PRC2 increases the sensitivity to EZH2 inhibition [2946].
Inhibition of histone demethylases may be an effective strategy to increase H3K27me3 levels. GSK-J4 is a small molecule inhibitor of KDM6, which removes the methyl groups from H3K27 and reduces H3K27me3 levels. This small molecule represses the growth of H3K27M DMG/DIPG cells and prevents colony formation in vitro, and it exhibits antitumor activity against K27M tumors in vivo [47]. Combination treatment with GSK-J4 and radiotherapy or the TP53-targeting/oxidative stress inducer APR-246 shows synergistic effects on cell death and restricts tumor growth in a xenograft mouse model [4849].
Although research has mostly focused on the H3K27me3 mark as the primary target of H3K27M, some studies have considered the potential role of increased levels of H3K27ac in tumorigenesis. Bromodomain and extra-terminal domain (BET) proteins are a family of proteins that bind to acetylated lysine residues on histones, particularly H3K27ac. JQ1, a BET inhibitor, represses tumor growth and extends survival in a DMG/DIPG xenograft model [29]. Another approach is to target the protein that transfers acetyl groups to H3K27. Histone deacetylase (HDAC) inhibitors can inhibit the growth of H3K27M DMG/DIPG cell lines, which have high levels of H3K27ac. Treatment of H3K27M cell lines using panobinostat, an HDAC inhibitor, increases H3K27ac levels but impairs cell viability, downregulating the proliferation-associated genes MKI67 and CCND1 in DMG/DIPG cells [31505152]. This small molecule shows synergistic effects with GSK-J4, JQ1, or the CDK7 inhibitor THZ1 in the treatment of DMG/DIPG [5052].
Targeting the altered metabolism of DMG/DIPG cells has emerged as a potential treatment strategy. A recent in vivo study showed that depleting MAT2A or methionine restriction could impede the growth of DMG/DIPG tumors. This suggests that DMG/DIPG tumors have a high dependency on methionine, and that targeting the methionine cycle could be a promising strategy for the treatment of this type of cancer [41].
The regulation of glucose is another therapeutic target. In DMG/DIPG, increased uptake of glucose upregulates the glucose transporter GLUT1, which is associated with tumor invasion. Treatment with the selective GLUT1 inhibitor BAY-876 suppresses tumor invasion and expansion without any side effects [36].
Another potential target for the treatment of DMG/DIPG is the regulation of alpha-ketoglutarate. A recent study published in Cancer Cell demonstrated that the H3K27M mutation in DMG/DIPG is associated with increased glycolysis, the tricarboxylic acid cycle, and glutaminolysis, which upregulates alpha-ketoglutarate and contributes to sustained epigenetic changes marked by H3K27me3 deficiency. Alpha-ketoglutarate is synthesized from isocitrate by isocitrate dehydrogenase1/2 (IDH1/2) or from glutamate by glutamate dehydrogenase (GDH). The inhibition of both pathways increases H3K27me3 levels and decreases tumor growth in vitro and in vivo, indicating that targeting these metabolic pathways is a promising strategy for the treatment of DMG/DIPG [39] (Table 2).
Blocking these metabolic and epigenetic pathways represents a promising strategy for the treatment of DMG/DIPG. However, further research is needed to determine the effectiveness of these approaches and their potential side effects.
CONCLUSION
DMG/DIPG, which was first documented in the literature in 1926, remains a significant challenge in the medical field [53]. Although approximately 150–400 new cases are diagnosed each year in the United States, little progress has been made in improving the prognosis and outcome of this tumor. Because the cause of DMG/DIPG remains unknown, radiation therapy is the only treatment available for this disease. In 2012, a unique histone 3.3 (H3-3A) K27M mutation found in most DMG/DIPG cases was identified by whole-genome sequencing; since then, research on this devastating disease has focused on the epigenetic changes associated with it. Recent research has shed light on the role of epigenetic changes in the development of DMG/DIPG. Specifically, it decreases an epigenetic mark called H3K27me3, which is normally present on histone H3 and represses the expression of certain genes. The loss of H3K27me3 in DMG/DIPG leads to a global shift in gene expression, and many genes that are normally silenced become active. This abnormal gene expression pattern, which includes metabolic genes, contributes to the development and progression of the tumor. The metabolic alterations observed in DMG/DIPG may serve as a novel diagnostic method. A recent study found that 11C-methionine PET imaging features with multiple biomarkers were correlated with progression-free survival and overall survival [54]. Also, ongoing research focuses on therapies that specifically target these mutated histones or the pathways they activate to improve the outcomes of DMG/DIPG patients. This will pave the way for the development of new therapies, including clinical trials for targeted molecular therapies. One of the leading groups in the field, that of Dr. Monje, established primary cells from DMG/DIPG patients and identified potential drugs using high-throughput screening [51]. There are currently 87 clinical trials across 133 countries focusing on DMG/DIPG (as of April 21, 2023; https://dipg.org/dipg-treatment/active-clinical-trials/). Additional research is needed to improve our understanding of the biology of this disease, which will help develop effective therapies to improve survival rates and the quality of life of children with DMG/DIPG.


 XML Download
XML Download