

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Transient receptor potential melastatin 4 (TRPM4) is a Ca2+-activated, nonselective monovalent cationic channel that does not allow the passage of divalent Ca2+ or Mg2+ ions [1,2]. Its expression is observed in various organs and tissues throughout the body [3-6]. TRPM4 plays a crucial role in regulating diverse cellular processes, including cell migration, proliferation, and differentiation [7-9]. Under normal physiological conditions, TRPM4 interacts with focal adhesion molecules involved in cell assembly and adhesion processes, thereby contributing to cell migration [10]. This interaction occurs in dendritic and immune cells, including T lymphocytes and mast cells, and contributes to cellular functions [11-15]. Due to its widespread expression and diverse functional roles in different cell types, TRPM4 has been implicated in the pathophysiology of various cardiovascular and immune diseases [16-18].
In recent years, research on TRPM4 has extended to its potential involvement in different types of cancer. Studies have shown that TRPM4 is overexpressed in several cancer cell lines, including HeLa cells (cervical cancer), as well as cells from prostate, colorectal, liver, breast, urinary bladder, and endometrial cancers [15,19-22]. The overexpression of TRPM4 has been associated with key cancer functions such as increased cell proliferation, migration, and alterations in the cell cycle.
To investigate the diverse functions of TRPM4 and gain a better understanding of its potential contribution to various pathophysiological conditions, it is essential to have a reliable tool or method for accurately detecting its expression. Additionally, understanding its trafficking and degradation can provide valuable insights into the regulation of TRPM4 and its involvement in various cellular processes.
Epitope tagging of TRPM4 is a useful strategy for studying its channel trafficking and expression. While previous studies have inserted an epitope at either the amino (N) or carboxy (C) terminus of TRPM4, this study suggests that the N-terminus is a more promising site for epitope insertion, as attempts to tag the C-terminus have been unsuccessful for other proteins like protein tyrosine phosphatase, non-receptor type 6 (PTPN6) [23]. To address this issue, we designed a mutant by inserting an epitope motif between the fifth (S5) and sixth (S6) transmembrane domains, allowing it to protrude from the cell membrane [24,25]. Epitope tagging has become a common approach for investigating various molecular aspects of proteins, including size, abundance, trafficking patterns, cellular localization, and other applications [26-29].
Considering the ease of insertion and detection, we selected a hemagglutinin (HA) tag, which is a nine-amino acid sequence (YPYDVPDYA) that shares two amino acids located in the extracellular region. This tag can be readily detected using a specific antibody without the need for membrane permeabilization with a detergent [30-32]. This strategy was adopted from previous studies on the heteromeric assembly of TRPC3 with TRPC4 [33]. However, it remains unknown whether the HA tag attached to TRPM4 disrupts its structure, expression, or channel function. Therefore, this study aims to determine whether TRPM4-HA can safely replace the endogenous protein and serve as a reliable marker in TRPM4-related research on pathophysiological conditions and diseases.
METHODS
Chemicals
The TRPM4 blockers 9-phenanthrol and N-Methyl-D-glucamine (NMDG) were purchased from Sigma–Aldrich.
Construction of HA-inserted TRPM4 and its YF mutants
cDNA encoding human TRPM4 (GenBank accession no. NM_017636) in EGFP-IRES vector was used to generate HA epitope-tagged TRPM4. For HA epitope tagging in the extracellular S6 domain of TRPM4 between amino acids Y1015 and A1016, the nucleotide sequence encoding HA (YPYDVPDYA) was incorporated into the primers by site-directed mutagenesis (EZchange Site-directed Mutagenesis kit; Enzynomics). The primers were designed based on the reported sequence of human TRPM4; Forward primer 5’-TTCCAGATTATGCCAACTGGCTGGTGGTGCTGCT-3’ and Reverse primer 5’-CATCGTATGGAGTACTGGGAGACGCAGGTGCCCG-3.’
The cytosolic PTPN6, interacts with and dephosphorylates TRPM4, thereby increasing the activity of TRPM4 [23]. To confirm whether PTPN6 is required for activation of TRPM4, pSicoR-PTPN6-shRNA was generated using oligonucleotide-directed mutagenesis (Enzynomics). Nucleotides 1148–1169 (5’-GGAGCATGACACAACCGAATA-3’) in human PTPN6 (GenBank accession no. NM_002831) were selected as a target shRNA. Human PTPN6-shRNA was validated by confirming the sequence and then cloned into adenoviral vectors using the Gateway cloning system (Thermo Fisher Scientific).
To confirm the interaction between PTPN6 and the tyrosine residues of TRPM4, phosphorylation-incompetent TRPM4-HA mutants were generated. Tyrosine phosphorylation sites were predicted using NetPhos-3.1 (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1). Four Tyr (Y) residues in the N-terminal region of TRPM4 (residues 75, 256, 393, and 519) were identified as putative phosphorylation sites. These four residues have been predicted as potential binding sites for PTPN6. Site-directed mutagenesis (Enzynomics) was performed to change the Tyr (Y) at residues 75, 256, 393, or 519 to Phe (F), which are referred to as YF mutants in the current study.
Cell culture and transfection
The HEK293 cell line was purchased from the Korean Cell Line Bank, and the cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FBS and 1% penicillin-streptomycin in an incubator at 37°C in 95% O₂ and 5% CO₂. The cells were transiently transfected with cDNAs using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s recommended protocol.
Subcellular fractionation and western blotting
HEK293 cells were transfected with TRPM4-HA or TRPM4-HA-YF mutants in the absence or presence of PTPN6-shRNA. After 48 h of incubation, the cells were harvested and lysed using a Membrane Protein Extraction Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The subcellular fractions were obtained by separating the cell membrane from the cytoplasm. The concentrations of solubilized proteins in the extracts were determined by BCA Protein Assay (Thermo Fisher Scientific). Proteins in the extracts (10 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The separated proteins were then transferred onto methanol-activated polyvinylidene difluoride membranes (Merck). The membranes were blocked with 5% skim milk in 1× TBST and washed three times for 10 min. The membranes were probed overnight with a primary rabbit antiserum against TRPM4 (1:1,000, Alomone) at 4°C and then washed three times for 10 min. The membranes were then incubated with horseradish peroxidase-labeled goat anti-rabbit secondary antiserum (1:1,000) (Thermo Fisher Scientific) for 1 h at room temperature.
Immunoreactive protein bands were detected with an iBright western blot imaging system (Thermo Fisher Scientific) using enhanced chemiluminescence reagents (Thermo Fisher Scientific; ratio of reagents A to B = 1:1). The same membrane was then stripped and probed with mouse primary antiserum against α-tubulin (1:1,000) (Thermo Fisher Scientific) to normalize the blots.
Immunocytochemistry
For immunocytochemistry, the HEK293 cells were seeded onto glass coverslips in 24-well plates. Once the cells reached 70%–80% confluency, transfection was conducted with TRPM4-IRES-GFP or TRPM4-HA-IRES-GFP. On the day of the experiment, the coverslips were washed in Dulbecco's phosphate-buffered saline (DPBS) and then fixed with 4% formaldehyde in PBS for 20 min at room temperature, followed by three washes in DPBS. To permeabilize the cells, the coverslips were incubated in 0.5% Triton X-100 in DPBS for 15 min. This step facilitates the removal of lipids from plasma and nuclear membranes, allowing the antibody to penetrate the cell and reach its interior. After extensive washing in DPBS, both permeable and non-permeable cells were incubated in a blocking solution (3% serum in DPBS) for 1 h. The cells were then further incubated with an anti-HA-tag monoclonal antibody (Invitrogen) diluted 1:500 for 1 h. After washing three times in DPBS, the coverslips were incubated with 1:325 diluted goat anti-mouse IgG H&L secondary antibody (Abcam) for 30 min. Then cells were washed in DPBS, the coverslips mounted with Fluorescence Mounting Medium (Dako), and then dried for 2 h in the dark. Images were captured using a confocal microscope equipped with a 405/488/594 nm laser for blue, green, and red fluorescent proteins (FLuoView; Olympus). The background intensity was normalized, and the images were analyzed using FLuoView software.
To determine the surface levels of TRPM4-HA and the YF mutants, HEK293 cells were seeded onto glass coverslips in 24-well plates. After 24 h, the cells were transfected with various types of TRPM4 cDNAs encoding IRES-GFP vector. The plasma membrane marker vector DsRed-Mem (RFP; Takara Korea Biomedical, Inc.) was used for co-transfection. Following transfection, the cells were incubated for a further 48–72 h and washed twice with PBS. The cells were fixed with 4% formaldehyde in PBS for 20 min at room temperature and then washed extensively. The cells were incubated with 4’,6’-diamidino-2-phenylindole (DAPI) (1:10,000, Thermo Fisher Scientific) solution for 5 min. After removal of the staining solution, the cells were washed three times with DPBS. Confocal fluorescence images were obtained, and the background intensity was normalized. Images were analyzed using FLuoView software.
Electrophysiology
For the electrophysiology experiments, a bath solution (140 mM NaCl, 5 mM CsCl, 1 mM CaCl2, 1 MgCl2, 10 mM glucose, and 10 mM HEPES, pH adjusted to 7.4 with NaOH) was used. A pipette solution (140 mM CsCl, 5 mM NaCl, 0.83 mM CaCl2, 1 mM MgCl2, and 1 mM EGTA, pH adjusted to 7.2 with CsOH) was used. For macro electrophysiology experiments, Bath 1 solution was prepared containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl₂, 10 mM glucose, and 10 mM HEPES (pH adjusted to 7.2 with NaOH), Bath 2 solution containing 140 mM NaCl, 5 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH adjusted to 7.2 with NaOH), and Bath 3 solution containing 140 mM NMDG, 5 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH adjusted to 7.2 with HCL). The solutions were applied using a gravity-driven local perfusion system. All the recordings were performed at room temperature.
Patch pipettes were prepared from borosilicate glass capillaries (Warner Instruments). Whole-cell or inside-out currents were recorded using a patch-clamp amplifier (Axopatch 200B; Molecular Devices). Current–voltage (I–V) relationships were measured by applying a linear ramp protocol of 400 ms from –100 to +100 mV. The holding potential was set to approximately –40 mV, which is similar to the resting membrane potential of most HEK293 cells. The sampling interval was 200 µs, and the data were filtered at 2 kHz. The currents were analyzed using Clampfit software (Molecular Devices). Currents at ± 100 mV were used to measure the current densities by subtracting the amplitude of the initial currents from the activated TRPM4 current and dividing it by the cell capacitance (pA/pF).
Currents in either whole-cell or inside-out configurations were completely desensitized within tens of seconds and then stabilized at the basal current (BC) level. After basal currents lasting for at least 1 min, 9-phenanthrol was applied for 3 min and washed. A Na+-free solution was prepared by substituting Na+ ions with NMDG. To determine the percentage of inhibition by 9-phenanthrol, the ratio between currents at +100 mV in TRPM4 and TRPM4-HA or TRPM4-HA-YF mutants was used.
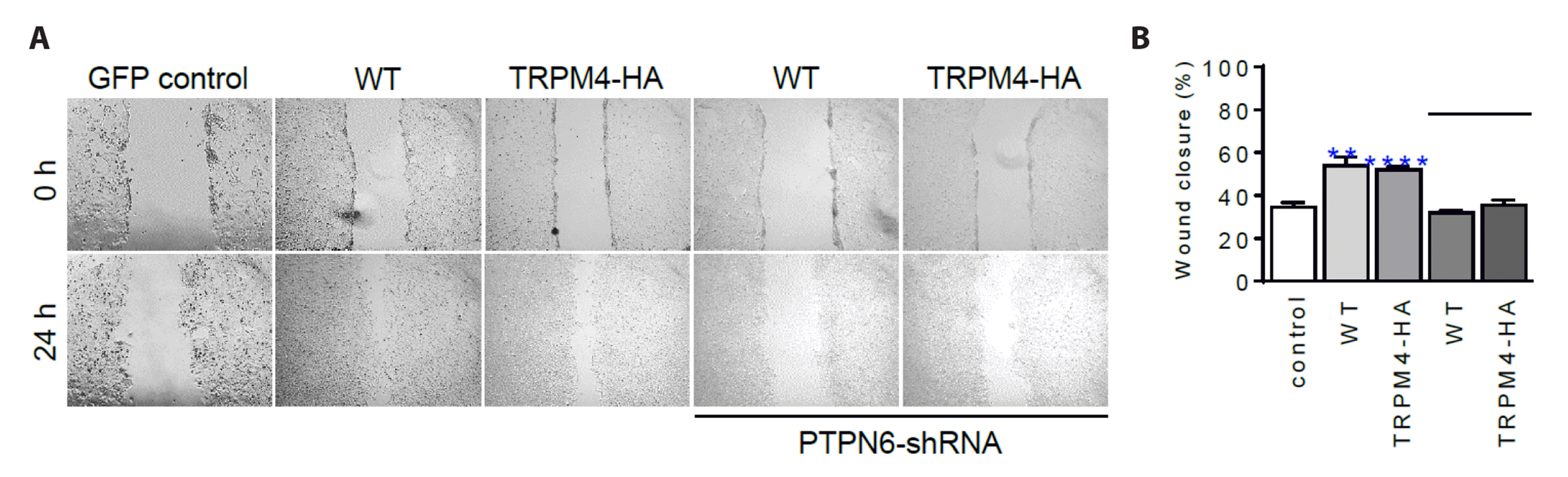
Cell proliferation assay (wound-healing assay)
For the wound-healing assay (WHA), HEK293 cells were cultured in 24-well plates and subsequently transfected with TRPM4-HA or TRPM4-HA-YF mutant cDNA. When the cells reached 90% confluency, a small scratch was created across the cell layer using a sterile 10 µl pipette tip. Images of the cells were captured at 0, 16, 24, and 40 h with a microscope (Carl Zeiss) to assess cell proliferation and migration.
Bimolecular fluorescence complementation (BiFC) assay
TRPM4-HA or its YF mutant was cloned into pBiFC-N-terminal VN (1–172) and PTPN6 into pBiFC-C-VC (173–273) vector. HEK293 cells were cotransfected with the two cloned BiFC vectors and incubated for 24 h. Following incubation, the cells were fixed with 4% paraformaldehyde for 20 min at room temperature and stained with DAPI (1:10,000, Thermo Fisher Scientific) for 5 min at room temperature. Coverslips were mounted with Fluorescence Mounting Medium (Dako), and Venus fluorescence signals were detected using a confocal microscope (Nikon A1).
RESULTS
The extracellular loop of TRPM4 was successfully tagged with HA-epitope
To assess the surface expression of TRPM4-HA, Western blotting and confocal microscopy with image analysis were conducted, as illustrated in Fig. 1. As anticipated, TRPM4-HA exhibited predominant expression in the membrane fraction, similar to the wild-type TRPM4 (WT). The expression of both WT and TRPM4-HA decreased when PTPN6 was silenced using its specific shRNA (PTPN6-shRNA in Fig. 1B), indicating that the trafficking and expression of TRPM4-HA, like WT, rely on PTPN6. In Fig. 1C, the presence of HA in the intact membrane confirmed successful exposure of the HA tag inserted in the extracellular loop, enabling binding with its specific antibody and fluorescent dye in the extracellular space.
The HA-tagged TRPM4 channel preserved the physiological characteristics of TRPM4
Supplementary Fig. 1 demonstrates that exogenous TRPM4 transfection increased the BC density at +100 mV by approximately three-fold, from 54.6 ± 11.0 pA/pF (GFP, n = 10) to 157.4 ± 18.83 pA/pF (TRPM4, n = 9), both in whole-cell configurations and excised inside-out patches (Supplementary Fig. 1B). Although variations in current density amplitude were observed due to differences in transfection efficiency, the response rates to chemicals remained relatively consistent, regardless of the amplitude variations (see Fig. 2 and Supplementary Fig. 1).
During recordings lasting approximately 20 min, the current levels exhibited a continuous decline (Fig. 2A). The current-voltage relationship displayed typical outward rectification for both TRPM4 (WT) and TRPM4-HA. In contrast, control cells exhibited a linear I-V relationship (Fig. 2B, C). The currents rapidly desensitized within a minute to a steady lower level, referred to as the BC, which was used as a scale for comparison with the WT.
Whole-cell currents recorded from cells transfected with TRPM4-HA were significantly inhibited by 9-phenanthrol (50.6 ± 10.2%, n = 7) at a similar rate as TRPM4-transfected cells (40.9 ± 5.7%, n = 8). In contrast, BC inhibition by 9-phenanthrol was insignificant in the GFP-transfected control cells (9.9 ± 4.6%, n = 10, see Fig. 2B).
Similar inhibition rates were observed in the inside-out configuration (57.3 ± 2.5% for TRPM4-HA, 58.6 ± 3.3% for WT). As shown in Fig. 2B, 9-phenanthrol had minimal inhibitory effects on the BC in GFP-transfected cells (Fig. 2C). When NMDG was applied instead of Na+, all the BCs obtained from cells expressing either WT or TRPM4-HA were reduced to their lowest levels (Supplementary Fig. 1C). Furthermore, when NMDG was applied to the bath solution (internal application in the excised inside-out configuration), the reductions in BCs were more pronounced at +100 mV (< 40%) compared to –100 mV (61%–79%) for both WT and TRPM4-HA. These findings confirm that the electrical properties of TRPM4-HA and WT were identical.
The expression of HA-tagged TRPM4 was found to enhance cell proliferation
Previous studies have reported that TRPM4 promotes cell proliferation and migration [34]. These functions were observed in cells expressing TRPM4-HA, as shown in Fig. 3. The evaluation time was set to 24 h after transfection, as depicted in Supplementary Fig. 2. Additionally, we investigated whether the knockdown of PTPN6 using specific shRNA (PTPN6-shRNA) affected the surface expression of TRPM4-HA (Supplementary Fig. 3) and suppressed cell proliferation (Fig. 3).
The proliferation rate, assessed by WHA, was found to be higher in groups transfected with WT (54.0 ± 4.1%, n = 6) and TRPM4-HA (52.0 ± 1.3%, n = 6) compared to the GFP control group (34.5 ± 2.2%, n = 6) (Fig. 3). However, when PTPN6 was silenced, indicated by the solid bar (PTPN6-shRNA), the proliferation rates at 24 h were reduced by approximately 31.7 ± 0.9% (WT, n = 6) and 35.5 ± 2.3% (TRPM4-HA, n = 7), similar to the control GFP group (Fig. 3B). These results support the idea that TRPM4-HA retains the functional properties of WT. Furthermore, the trafficking of TRPM4-HA was found to be dependent on PTPN6, similar to WT [23].
The investigation focused on the impact of tyrosine phosphorylation in the N-terminus using TRPM4-HA
The aforementioned results demonstrated that the properties of TRPM4 were unaffected by the addition of the HA tag, even when PTPN6 was knocked down. Since PTPN6 is necessary for the trafficking of TRPM4, this study aimed to identify the specific tyrosine (Y) residue in the N-terminus responsible for PTPN6 binding, based on previous findings indicating the exclusive binding site for PTPN6 is the N-terminus [23]. Four Y residues, namely Y75, Y256, Y393, and Y519, predicted to be involved in tyrosine phosphorylation, were selected and mutated to phenylalanines (F) using TRPM4-HA as the framework.
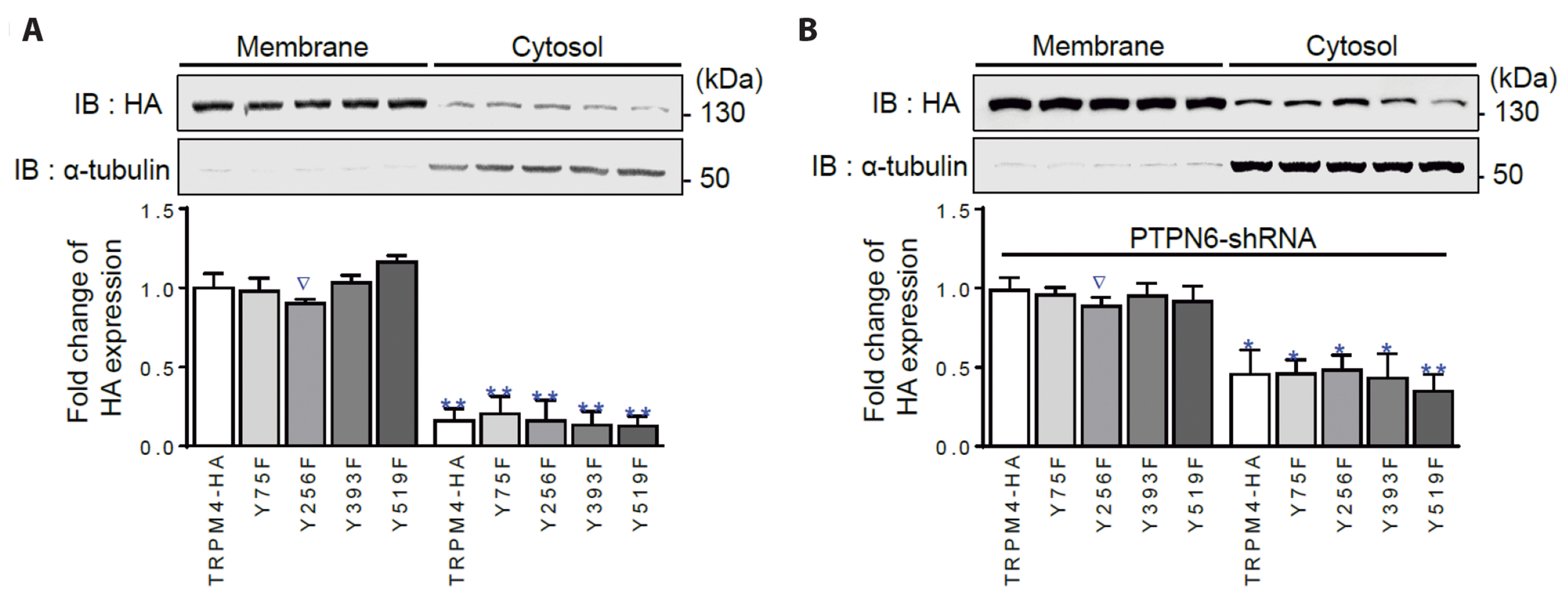
Initially, the expression of the four YF mutants (Y75F, Y256F, Y393F, and Y519F) in the membrane was examined. All four YF mutants were expressed in the membrane, as shown in Fig. 4A and Supplementary Fig. 3. The Y256F mutant exhibited a slightly lower expression tendency compared to the other YF mutants (∇ in Fig. 4), although this difference was not statistically significant. PTPN6 knockdown using a specific shRNA increased the retention of the mutants in the cytosolic fraction by more than two-fold, similar to the control TRPM4-HA (Fig. 4B). To confirm that the tested Y residues did not interact with PTPN6, a BiFC assay was performed. All four YF mutants interacted with PTPN6, similar to TRPM4-HA, without any statistically significant differences between the WT and mutants (Supplementary Fig. 4).
To investigate whether the TRPM4-HA-YF mutations affected the electrical properties of TRPM4, current densities were measured in cells expressing TRPM4-HA and TRPM4-HA-YF mutants. The current densities ranged from 180 to 266 pA/pF (n = 7–13) for both TRPM4-HA and TRPM4-HA-YF mutants (Fig. 5A), while the current densities in the inside-out patch configuration were in the range of 10–21 pA/pF (n = 6–16) (Fig. 5B). However, the inhibition rate of 9-phenanthrol in the TRPM4-HA-YF mutants was approximately half of that in TRPM4-HA, ranging from 22% to 45% (n = 8–13) (Fig. 5). In the inside-out patch configuration, the responses to 9-phenanthrol were also more resistant, ranging from 21% to 33% (n = 9–16), compared to TRPM4-HA (57.3 ± 2.5%, n = 6). Among the TRPM4-HA-YF mutants, the Y256F mutation exhibited the most significant decrease in the inhibition rate (summary bar charts in the right panel, Fig. 5A: 21.3 ± 4.6%, n = 12; Fig. 5B: 20.8 ± 3.9%, n = 9).
As depicted in the summary in Fig. 6, the results obtained from the WHA assay demonstrated that the TRPM4-HA-YF mutants significantly boosted cell proliferation within the range of 72.6% to 79.7% (n = 8 for each mutant) compared to the negative control (N/C). However, TRPM4-HA exhibited the highest level of proliferation at 90.1%. In contrast, the N/C group displayed only half the proliferation rate (51.6 ± 2.5%, n = 8) compared to both the TRPM4-HA-YF mutants and TRPM4-HA. When compared to non-treated naive cells (51.6 ± 2.5%, n = 8, N/C), cells with TRPM4-HA-YF mutations showed increased cell proliferation and migration ranging from 72.6 ± 3.0% to 79.7 ± 4.0% (n = 8 for each TRPM4-HA-YF mutation). However, these values were lower than those observed in the TRPM4-HA group (90.1 ± 2.8%, n = 8) (Fig. 6A). Under conditions of PTPN6 knockdown, the wound closure rates for all groups were delayed by half (50.2%–53.6%, n = 8 each) (Fig. 6B). These results indicate that the four TRPM4-HA-YF mutations are unlikely to directly participate in cell proliferation and migration.
DISCUSSION
The primary focus of the TRPM4 channel investigations has been on changes in its expression and membrane translocation. To address this, we developed a mutant variant, TRPM4-HA, which allows for the monitoring of TRPM4 trafficking in intact cells. Our aim was to assess whether its properties are comparable to those of the native TRPM4 and whether it can be utilized effectively and safely, similar to TRPM4.
Using the recently reported structure of TRPM4 [24,35], we constructed the TRPM4-HA mutant by incorporating an HA epitope. This involved inserting additional sequences before the endogenous YA located in the extracellular loop extending outside the cell. This approach preserves the integrity of the membrane, mimicking physiological conditions and minimizing the risk of overestimation or misinterpretation due to contaminants in the cytoplasmic and membrane fractions.
Detection of the epitope in the intact membrane confirmed the stable presence of the HA tag extending outward from the cell, indicating successful trafficking and surface expression of TRPM4-HA. Importantly, TRPM4-HA exhibited channel properties identical to TRPM4, as demonstrated by electrophysiology and pharmacology experiments. Furthermore, the cell proliferative function and dependence on PTPN6 for trafficking were conserved with this mutant, mirroring those of TRPM4. These results establish that TRPM4-HA functions as a TRPM4 channel with an extracellular HA epitope.
Investigations using TRPM4-HA and its YF mutants did not provide clear information about the binding site with PTPN6, which is crucial for TRPM4 trafficking. Interestingly, the TRPM4-HA-YF mutants showed reduced sensitivity to 9-phenanthrol compared to TRPM4-HA. Among these mutants, Y256F exhibited the lowest sensitivity, suggesting that Y256 is likely part of the 9-phenanthrol binding site. Y256 is located in the α6 helix, which forms part of the bottom of the N-terminal TRPM4 homology regions 1 and 2 (MHR1/2). These regions are positioned beneath the C-terminal domain and may play a role in gating and ion channel activities [24]. Therefore, Y256 within MHR1/2 is a potential site for 9-phenanthrol to modulate ion channel activity. Additionally, the Y256F mutant appeared to resist membrane translocation, possibly contributing to lower channel currents. However, since the current sizes showed little variation, it is more likely that the studied Y residues all play a role in modulating channel function.
BiFC analysis targeting the four residues predicted by tyrosine phosphorylation did not provide evidence of protein-protein interactions between PTPN6 and each YF mutant. This suggests that PTPN6 stably binds to the non-phosphorylated form of TRPM4. However, this study examining the selected Y residues could not determine whether PTPN6 directly dephosphorylates TRPM4 or identify the specific site(s) responsible for this effect. The examination under PTPN6 knockout conditions in this study did not yield direct information about PTPN6's effect on tyrosine phosphorylation.
In conclusion, TRPM4-HA fulfills the requirements for investigating TRPM4, serving as a valuable tool for identifying critical structures involved in pathway expression and migration, thanks to the internal HA tag in the extracellular loop. TRPM4-HA is particularly well-suited for studying cell functions related to morphological changes and expression patterns in relation to TRPM4.
SUPPLEMENTARY MATERIALS
Supplementary data including four figures can be found with this article online at https://doi.org/10.4196/kjpp.2023.27.4.417.

 XML Download
XML Download