

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Medulloblastoma (MBL), one of the central nervous system (CNS) embryonal tumors arising from the cerebellum, is the most common pediatric malignant brain tumor. MBL is currently treated with maximal safe resection, chemotherapy, and craniospinal irradiation (CSI). With multimodal treatment and appropriate risk stratification, the long-term survival rate has improved in 70%–80% of all patients. Despite aggressive multimodal therapy, approximately 30% of patients eventually succumb to this disease, and survivors cope with the long-term side effects of treatment that have a significant impact on their quality of life. Recent molecular studies have demonstrated the clinical and biological heterogeneity of MBL. MBL is classified into at least four subgroups: wingless (WNT), sonic hedgehog (SHH), group 3, and group 4. Previously, MBL has been stratified by age at diagnosis, metastatic status, tumor removal extent, and presence or absence of large cell/anaplastic (LC/A) histology. In this review, the epidemiology, diagnosis, clinical manifestation, classification, and current treatment strategies based on recently published studies are presented.
EPIDEMIOLOGY
CNS embryonal tumors are the most common group of malignant CNS tumors, which represents 20% of pediatric CNS tumors [123]. MBL comprises 15% of all pediatric CNS tumors, 40% of all posterior fossa (PF) tumors, and 90% of all embryonal tumors. MBL accounts for most embryonal tumors arising from the PF. The incidence in ages 0–19 years is 0.41 cases per 100,000 patient-years and decreases with age [4]. The peak incidence is between 5 and 7 years of age and occurs more frequently in males, with a male:female ratio of 1.7:1 [5].
DIAGNOSIS AND CLINICAL MANIFESTATION
The diagnosis of MBL is based on clinical symptoms and imaging findings, such as CT scan and MRI. MBL commonly presents with headaches (especially morning headaches), nausea/vomiting, lethargy, and ataxia, resulting from increased intracranial pressure and cerebellar dysfunction [6]. After surgery, a follow-up MRI is recommended to determine the size of the residual tumor. Histopathologic confirmation and classification are performed using integrated histopathology and molecular studies. Spine MRI is often done preoperatively to evaluate metastatic disease. If preoperative spine MRI was not performed, it is recommended to perform MRI more than 2 weeks after surgery to avoid false positives results [7]. To confirm cerebrospinal fluid (CSF) dissemination at diagnosis, CSF cytology tests are usually done 2–4 weeks after surgery [8]. The differential diagnosis of MBL is atypical teratoid/rhabdoid tumor, ependymoma, pilocytic astrocytoma, and choroid plexus papilloma.
CLASSIFICATION
According to the WHO 2021 CNS5 classification, MBL is molecularly classified into four groups: 1) WNT-activated, 2) SHH-activated and TP53-wildtype, 3) SHH-activated and TP53-mutant, and 4) non-WNT/non-SHH. In addition, MBL is histologically divided into four types: 1) classic, 2) desmoplastic/nodular (DN), 3) extensive nodularity (MBEN), and 4) LC/A [9]. It was updated to one category in the WHO 2021 CNS5 classification as “medulloblastoma, histologically defined” [10]. SHH MBL has been primarily associated with DN and MBEN histology [11]. LC/A MBL is commonly observed in SHH TP53-mutant and infant group 3 [12]. Classic MBL is observed in nearly all WNT and most groups 3 and 4 [13].
As molecular subgroups have been incorporated into the WHO classification, more molecular tools are needed for precise classification. Modern diagnostic pathology for CNS tumors uses DNA/RNA analysis, methylome profiling, and microscopy and focuses on diagnostic and prognostic markers [14]. Molecular tools aid in classifying tumors, inform about the natural history of tumors, and advise about the probability of response to specific therapeutic regimens. Table 1 presents the key genes and proteins required for integrated diagnosis.
MOLECULAR SUBGROUPS
MBL comprises four molecular disease subgroups, including WNT, SHH, group 3, and group 4, by consensus meeting [13]. Each group was defined based on genome-wide transcriptomic and methylomic signatures.
WNT (wnt/wingless pathway) MBL
The WNT subgroup accounted for approximately 10% of all MBL. WNT MBL usually develops in the midline cerebellum and may spread to the dorsal brainstem [15]. It usually presents with classic histology, and metastasis is rare at diagnosis, occurring in less than 5% of patients [12]. WNT MBL is characterized by mutations that cause constitutive activation of the WNT signaling pathway [16]. Approximately 85%–90% of patients with WNT MBL have a somatic activating mutation in exon 3 of CTNNB1. This stabilizes β-catenin and induces sustained activation of the WNT pathway [17]. Germline APC mutations are also associated with WNT MBL. Another hallmark of WNT MBL is monosomy 6, which usually coexists with a CTNNB1 mutation [18]. Other frequently mutated genes were DDX3X, SMARCA4, and CREBBP. Childhood WNT MBL (<16 years of age at diagnosis) shows a favorable prognosis, with a 5-year survival rate of more than 90%.
SHH MBL
The SHH subgroup is most common in infants and young adults, accounting for 25% of all MBL. SHH MBL commonly develops in the cerebellar hemispheres; however, some also occur in the midline [15]. It shows mutations or copy number alterations of genes related to the SHH signaling pathway [19]. Loss-of-function mutations or deletions in PTCH1 and SUFU and activating mutations in smoothened homolog (SMO), GLI1, and GLI2, as well as MYCN amplification, may be present. The hallmark cytogenetic events in SHH MBL include loss of chromosomes 9q and 10q, which induces loss of heterozygosity of PTCH1 and SUFU. TP53 mutations are associated with poor outcomes in patients with SHH MBL. TP53 loss-of-function mutations may co-occur with clustered chromosomal rearrangements, known as chromothripsis, with MYCN and/or GLI2 amplification [20].
Robinson et al. [21] revealed that infant SHH could be classified into SHH-I and II, and SUFU aberrations and chromosome 2 gain were enriched in infant SHH-I. Another study suggested that the SHH group is divided into α, β, γ, and δ, and infant SHH-I and II correspond to SHH-β and SHH-γ, respectively [22]. The prognosis of SHH-γ (infant SHH-II) is better than that of SHH-β (infant SHH-I).
Groups 3 and 4 MBL
Groups 3 and 4 MBL are heterogeneous with some degree of molecular overlap and frequently arise in the midline, occupying the fourth ventricle [15].
Group 3 MBL
Group 3 comprised 25% of MBL, mainly in infants and young children. Outcomes are inferior to those of other subgroups, with a survival rate of 40%–60%. Metastatic disease at diagnosis appears in 40%–45% of patients. LC/A histology (40%) and high-level MYC amplification are features of group 3 MBL (17%) [23]. Common chromosomal structural alterations include gains in 1q, 7, and 17q and deletions in 10q, 11, 16q, and 17p. Aberrant activation of GFI1 and GFI1B by enhancer hijacking is observed in 15%–20% of group 3 MBL [24]. MYC amplification and isochromosome 17q are associated with poor prognostic biomarkers.
Group 4 MBL
Group 4 MBL is the most common subgroup, comprising 35% of cases, and occurs across the age spectrum. Group 4 MBL is commonly driven by enhancer hijacking-mediated PRDM6 overexpression (17%), associated with a focal tandem duplication of SNCAIP [19]. Gain of chromosomes 7 and 17q and deletion of chromosomes 8, 11, or 17p are common. Isochomosome 17q (80%) is the most common cytogenetic aberration but does not help predict survival. MYCN amplification and overexpression are common but are not associated with poor outcomes, such as SHH. As it is recently known that the prognosis is good for patients with chromosome 11 deletions, future Children’s Oncology Group (COG) studies are planning to lower the CSI dose for these patients [2526].
Recently, a new classification was published for groups 3 and 4 MLB by analyzing 1,501 patients in groups 3 and 4 MBL cohorts. Groups 3 and 4 MBL are newly classified as types I–VIII by recent high-resolution subclassification approaches. Subtypes I, V, and VII were mixed with groups 3 and 4. Subtype III, classified as high-risk, is characterized by MYCC/MYCN amplification. Subtype VII is characterized by KBTBD4 mutation. Each type has different driver events and cytogenetics and differs in terms of survival [222728].
TREATMENT
Current treatment for MBL consists of maximal safe resection, chemotherapy, and CSI. The surgical standard is maximal safe resection which was established based on several studies supporting the relationship between resection extent and survival rate [293031]. Aggressive resection where high neurologic morbidity is expected, such as when the brainstem is involved, is not recommended. Previously MBL was treated with 36 Gy of post-operative CSI with a boost of 54–55.8 Gy to the PF after surgery because of the radiosensitivity of the tumor. Since the 1990s, several studies have reported that adjuvant chemotherapy has improved the outcomes of MBL [32333435]. The factors that determine the prognosis of MBL are the extent of CNS disease at diagnosis, age at diagnosis, amount of residual disease after surgery, tumor histopathology, and biological and molecular tumor cell characteristics. This section describes the treatment strategies and outcomes according to the risk groups and strategies for optimizing risk stratification.
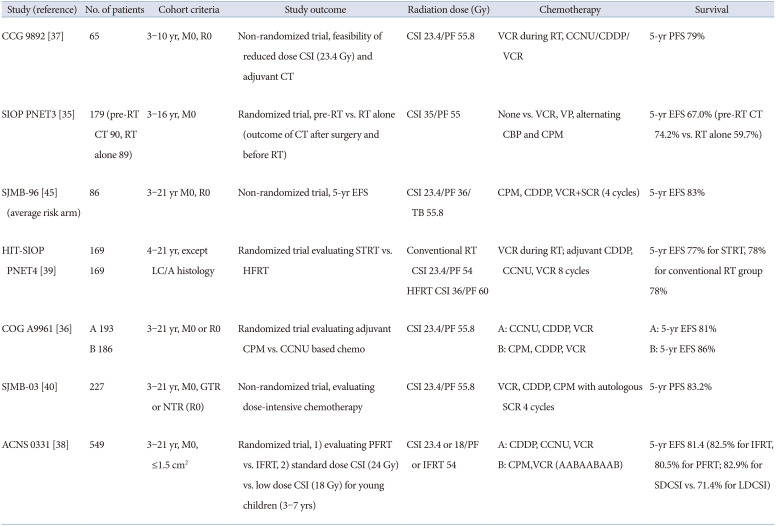
Older children–average risk
Average risk (AR) MBL was defined as non-metastatic MBL achieving gross total resection (GTR) not belonging to the LC/A histological subtype without MYC or MYCN amplification. Packer et al. [3637] showed excellent outcomes with a reduced dose of CSI (23.4 Gy) in young children (3–7 years) AR MBL due to long-term neurologic, cognitive, and endocrinologic sequelae of high-dose CSI. Therefore, the current treatment of AR MBL is CSI to 23.4 Gy with PF or involved field (IF) boost to 54 Gy followed by chemotherapy. Studies have also been conducted to determine which chemotherapeutic agents are effective. In the COG A9961 study, a randomized trial comparing chemotherapy based on cyclophosphamide, cisplatin and vincristine vs. lomustine (CCNU), cisplatin and vincristine was performed. There was no significant difference in survival between cyclophosphamide- and CCNU-based regimens [36].
Subsequently, further efforts have been made to lower the radiation therapy (RT) dose and volume. In a phase III randomized trial, a comparison between a smaller boost (radiation to the tumor bed) and a standard volume boost (radiation to the entire PF) was conducted. In addition, young children (3–7 years) were randomly assigned to receive 23.4 Gy vs. 18 Gy CSI. The involved field radiation therapy (IFRT) was deemed non-inferior compared with that of posterior fossa radiation therapy (PFRT). Unfortunately, children receiving low-dose (LD) CSI showed inferior event-free survival (EFS) compared with those receiving standard-dose (SD) CSI (71.4% vs. 82.9%, p=0.028) [38]. There was no significant difference in outcomes between LD and SD CSI in the WNT, SHH, and group 3. However, group 4 patients receiving LD CSI showed worse EFS than those receiving SD CSI [38]. In the HIT-SIOP PNET 4 trial, hyperfractionated (HF) vs. conventional RT followed by maintenance chemotherapy was evaluated. Hyperfractionated radiation therapy (HFRT) did not improve survival in AR MBL [39]. The St Jude medulloblastoma (SJMB)-96 and -03 trials investigated risk-adapted radiotherapy with 23.4 Gy CSI for patients with AR MBL. Through risk-adapted radiotherapy followed by short, dose-intensive, alkylator-based chemotherapy, the 5-year EFS for patients with AR MBL was 82%–83% in SJMB-96 and -03 trials [40]. The clinical trials for AR MBL are summarized in Table 2.
Recently, the survival rate of AR MBL has been reported to be more than 80%. According to the molecular subgroup, the outcome was 5-year EFS 93%–98% for WNT, 75%–83% for SHH, 63%–67% for group 3, and 86%–87% for group 4 [3840]. The WNT subgroup had the best outcome, while group 3 had the worst outcome. The Current SIOP PNET 5 MBL trial has five different arms: WNT-activated MBL as low-risk, non-WNT MBL as standard risk, WNT-activated MBL with high-risk features, and SHH-activated MBL with biologically very high-risk features (SHH-activated, TP53 mutated). Efforts to lower the treatment intensity for the WNT subgroup are described below in ‘future risk stratification’ section.
Older children - high risk
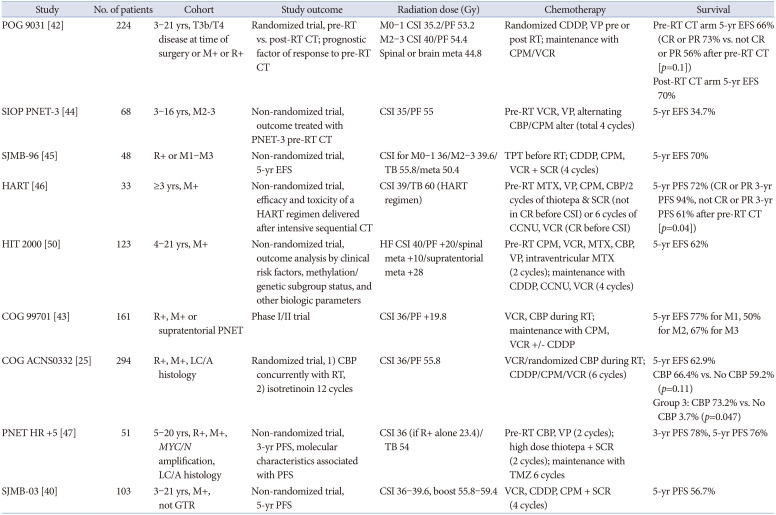
In current practice, high risk (HR) MBL is currently defined as having one or more of the following clinical factors: metastatic disease (Chang stages M1–M4; M+), LC/A histology, MYC or MYCN amplification, or significant residual disease after surgery (>1.5 cm2; R+) [41]. Approximately 30% of patients have metastases, and they have a poor prognosis. In the Pediatric Oncology Group (POG) 9031 study, M4 disease showed a significantly lower 5-year EFS than that of M0–M3 disease (70% vs. 22%) [42]. There is some controversy as to whether the presence or absence of residual tumors predicts a poor prognosis. Recent studies have reported that the extent of residual tumors does not affect the prognosis [3543].
A consensus on treatment for HR MBL has not yet been established. Currently, the standard dose of RT for HR MBL contains a dose of 36–39.6 Gy CSI and a boost of up to 54 Gy to the primary site. In the POG 9031 study, the 5-year EFS of the entire HR cohort improved to 68.1%. The relatively good outcome for M2–M3 disease (5-year EFS according to M stage: M0 74.0%, M1 64.9%, M2 69.2%, M3 61.6%, and M4 22.2%) could be attributed to the higher dose of CSI (40 Gy) [42]. However, the results of the SIOP/UKCCSG PNET-3 study, which applied pre-RT chemotherapy and RT to patients with M2–M3 MBL, were not satisfactory, suggesting that more intensive treatment is needed for HR MBL [44].
In phase I/II study of M+ MBL (COG 99701), carboplatin RP2D, as a radiosensitizer, was determined to be 35 mg/m2/dose ×30, suggesting that this could be a good strategy for M+ MBL [43]. Carboplatin may enhance the production and persistence of single- and double-strand breaks in DNA. Subsequent COG ACNS0332 was a randomized phase III study evaluating carboplatin concurrently with RT in M0 with >1.5 cm2 residual, M+, or diffusely anaplastic MBL, regardless of M-stage or residual tumor. As the 5-year EFS was 66.4% with carboplatin and 59.2% without carboplatin (p=0.11), concurrent carboplatin did not significantly improve the EFS in all patients. However, the efficacy was proven in the group 3 subgroup (73.2% with carboplatin, 53.7% without carboplatin, p=0.047). Therefore, concurrent carboplatin treatment during radiotherapy is recommended for pediatric patients with HR group 3 MBL [25].
High-dose chemotherapy and autologous stem cell transplantation (HDC/ASCT) have been applied in the SJMB-96, SJMB-03, HART, and PNET HR trials. The SJMB-96 study reported that a short, dose-intense, alkylator-based chemotherapeutic regimen was helpful in improving the outcome of HR MBL [45]. Gandola et al. [46] reported a 5-year progression-free survival (PFS) rate of 72 % with intensive postoperative chemotherapy, HFRT, and HDC/ASCT. In PNET HR+5, the 5-year PFS was 76% by applying two courses of pre-RT chemotherapy, two courses of high-dose thiotepa and ASCT, and maintenance temozolomide in HR MBL [47]. In Korea, a strategy of implementing reduced-dose craniospinal radiotherapy (CSRT) and applying tandem HDC/ASCT has been attempted [4849]. Sung et al. [49] reported that reducing the CSRT dose (23.4 or 30.6 Gy) and applying tandem HDC/ASCT (carboplatin-thiotepa-etoposide and cyclophosphamide-melphalan regimen) did not reduce survival (5-year EFS 70%) in HR MBL. Clinical trials for HR MBL are summarized in Table 3.
As shown in a previous study, the survival rate of HR MBL was improved by up to 70%. Histologically, DN MBL has the best prognosis, LC/A MBL has the worst prognosis, and the classic type has an intermediate prognosis (5-year EFS, 89% for DN, 61% for classic, and 40% for LC/A MBL in the HIT 2000 study). According to the molecular subgroup, 5-year EFS or PFS was 92%–100% for WNT, 25%–60% for SHH, 40%–60% for group 3, and 65%–68% for group 4 among clinical trials for HR MBL [254050].
The current SIOP-Europe HR MBL trial recruits patients with any HR factor (M+, LC/A pathology, MYC amplification, MYCN amplification, or TP53 somatic mutation [both in SHH tumors only]) from February 2021. They compared conventional radiotherapy vs. HF/accelerated radiotherapy vs. HDC with thiotepa, followed by conventional radiotherapy. They also compared two different maintenance chemotherapies, vincristine, CCNU, and cisplatin alternative with vincristine, and cyclophosphamide vs. temozolomide [51].
Infant MBL
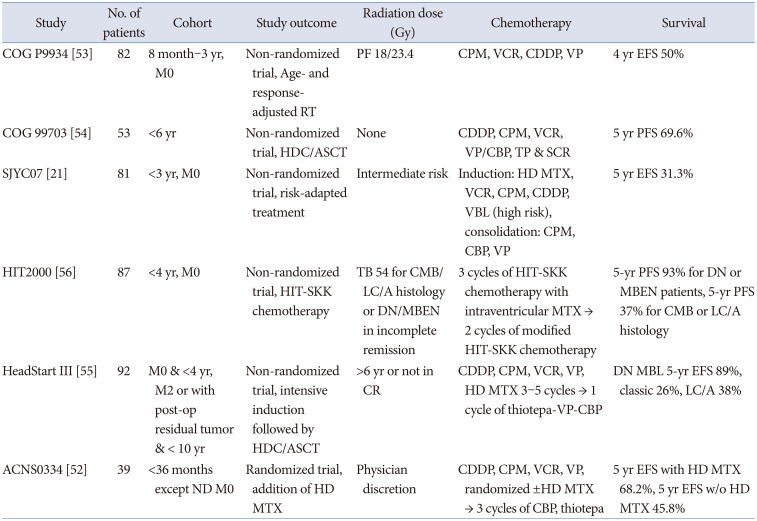
Limitations in the use of RT due to the high vulnerability of the developing brain and the high dissemination rate at diagnosis have resulted in a low survival rate in infant MBL. Several alternative treatments for avoiding RT have been attempted, including intraventricular or high-dose methotrexate (HD MTX), HDC/ASCT, and primary focal site RT [52].
The COG P9934 trial tested focal RT for PF with 18 or 23.4 Gy [53]. The COG 99703 study demonstrated a 5-year PFS of 69.6% and 5-year OS of 76.1% with induction chemotherapy (cisplatin, cyclophosphamide, vincristine, and etoposide) and three cycles of HDC (carboplatin and thiotepa) and ASCT [54]. The HeadStart III trial also used 3–5 cycles of adjuvant chemotherapy (cyclophosphamide, vincristine, etoposide, and HD MTX) and one cycle of HDC (thiotepa, etoposide, and carboplatin) and ASCT [55]. Carboplatin, thiotepa, etoposide, and busulfan are commonly used in HDC regimens. In the HIT 2000 study, systemic chemotherapy, intraventricular MTX, and risk-adapted local RT were applied to avoid CSI instead of HDC/ASCT in patients with non-metastatic MBL younger than 4 years of age [56]. The prognostic factors were M2 or higher, molecular subgroups, and histology. The COG ACNS0334 trial evaluated the role of HD MTX in young children aged under 3 years at diagnosis. After surgery, three cycles of induction chemotherapy were administered, including cisplatin, cyclophosphamide, vincristine, etoposide, and ±HD MTX in a randomized manner, followed by three cycles of consolidation (carboplatin, thiotepa) and ASCT. In preliminary data, the arm with HD MTX showed better 2-year EFS than that of the arm without HD MTX, but the difference was not statistically significant. The contribution of HD MTX was prominent in group 3 (5-year overall survival of 80% with HD MTX vs. 40% without HD MTX) [52]. Clinical trials on infant MBL are summarized in Table 4.
In the infant group, the SHH group showed a higher survival rate than that of group 3. DN/MBEN showed more than 90% of 5-year PFS, but non-DN/MBEN showed a lower 5-year PFS of less than 60% [54]. DN/MBEN MBL showed higher rate of GTR than classic MBL. In multivariate analysis, incomplete resection (GTR vs. non-GTR) and metastases were independent prognostic factors in young children [57]. This implicates that maximal safe resection is the reasonable strategy for the treatment of MBL. The ongoing HeadStart 4 trial investigates treatment reduction from two cycles to a single cycle of HDC/ASCT in children younger than 6 years with SHH MBL, TP53 wild-type, regardless of metastasis or extent of resection (NCT02875314) [52].
New approaches to incorporate subgroups into management
Currently, it is accepted that the WNT subgroup aged under 16 years is regarded as a low-risk group because of its favorable prognosis. In the WNT subgroup, the difference in the EFS according to the traditional HR and AR categories is not significant. Therefore, efforts are being made to lower the intensity of treatment because the treatment results of the WNT group aged under 16 years are good. Prospective clinical trials currently being conducted for the WNT group include SIOP-PNET5 (NCT02066220), ANCS1422 (NCT02724579), and SJMB-12 (NCT01878617) [5859]. In these studies, attempts to reduce the CSI dose are commonly applied (Table 5).
Although infant SHH MBL with DN histology usually shows favorable outcomes, SHH MBL with TP53-mutant and MYCN amplification shows disappointing survival. Therefore, novel treatment strategies are needed. In systematic review of phase I and II clinical trials, SMO inhibitors showed 37% and 0% of objective response rate in recurrent SHH and non-SHH MBL, respectively [60]. Based on these evidence, vismodegib, an SMO inhibitor, has been evaluated in newly diagnosed SHH MBL (>12 years) (NCT01878617).
Immunotherapy has been limited in MBL due to the lack of immunogenic antigens, tumor microenvironment, and blood-brain barrier. However, many efforts have been made to overcome limitations [26]. Early-phase clinical trials of chimeric antigen receptor T-cell therapy targeting NKG2DL, GD2, HER2, EGFR, G7-H3, natural kille (NK) cells, and dendritic cell therapy are also ongoing [6162].
FUTURE RISK STRATIFICATION
MBL was initially classified into four molecularly distinct subgroups and was further refined into 12 subtypes [19]. Prospective studies in which risk stratification was classified according to age, extent of resection, and metastasis also applied sub-analysis according to molecular subgroup and suggested future risk stratification. For example, the COG ACNS 0332 study analyzed metastatic WNT MBL as a favorable risk and group 3 with MYC or MYCN amplification as a very high-risk. In addition, MBL with chromosome 11 loss or chromosome 17 gain had superior survival in group 4 with 91.7% of 5-year EFS. They suggested therapy reduction in the WNT subgroup with metastatic disease and the group 4 subgroup with chromosome 11 loss and/or chromosome 17 gain [25]. In the SJMB-03 trial, WNT, low-risk SHH, low-risk groups 3 and 4, which are low groups with 5-year PFS expected to exceed 90%, and HR SHH, HR combined groups 3 and 4, which are very high-risk groups with 5-year PFS expected to be <60% were classified [40]. Accordingly, the combined groups 3 and 4 may be classified as low risk as M0 and subtype VII; intermediate risk as M0 and subtypes I, II, IV, V, VI, and VIII; and HR as M disease or subtype III or MYC amplification. However, this classification is not definitive and should be more accurately verified in future prospective clinical trials.
GERMLINE PREDISPOSITION
MBL has been associated with rare germline predisposition syndromes, including Gorlin syndrome (SUFU and PTCH1) [63], Li-Fraumeni syndrome, APC-associated polyposis syndromes [64], and Fanconi anemia [65]. Waszak et al. [66] reported that 6% of patients with MBL had germline mutations; APC, BRACA2, PALB2, PTCH1, SUFU, and TP53 were highly associated with MBL. WNT MBL is known to be related to germline APC mutations; germline APC and somatic CTNNB1 mutations are mutually exclusive. Therefore, if there is no somatic CTNNB1 exon mutation in WNT MBL, APC testing is recommended. All SHH tumors should be screened for somatic and germline TP53 mutations after appropriate genetic counseling. SUFU and PTCH1 mutations are also highly associated with SHH MBL. PALB2 and BRCA2 could be found in SHH and groups 3 and 4 MBL.
LATE EFFECTS
Patients with MBL have a high incidence of treatment-related secondary neoplasms and physical, neurocognitive, endocrine, and auditory late sequelae [6768]. Growth hormone deficiency and primary hypothyroidism are the most common endocrinologic complications [68]. Children receiving CSI have a greater risk of neurocognitive effects such as working memory, processing speed, and fine-motor functioning [69]. To reduce neurotoxicity, reduced-dose CSI, a smaller boost to the tumor bed, HFRT, and proton therapy have been attempted. COG developed a standard neuropsychological and behavioral battery to measure intelligence, processing speed, attention, memory, language preference, behavioral/social/emotional function, executive function, adaptive function, and quality of life [70]. Therefore, it is necessary to detect the late effects early and provide appropriate interventions.
CONCLUSION
Noteworthy progress has enabled us to better understand the clinical, biological, and molecular characteristics, resulting in biology-based risk stratification and tailoring treatment intensity to disease risk. Ongoing research focuses on careful treatment reduction in low-risk patients and novel therapies in high-risk patients. Ultimately, it should be in the direction of improving the quality of life while increasing the survival rate of patients with MBL.


 XML Download
XML Download