

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The antidiabetic effect of rosiglitazone, an oral thiazolidinedione (TDZ)-based antidiabetic drug, is mediated by activation of peroxisome proliferator-activated receptor γ and reduces blood glucose and glycated hemoglobin levels [1,2]. Rosiglitazone was once removed from the market because of its cardiac toxicity, and then the US Food and Drug Administration (FDA) lifted restrictions on its use. Since then, the use of this drug has been gradually increasing based on reports that it improves insulin resistance along with other TDZ-based diabetes drugs [3].
One of the Shaw-type K+ channels, Kv3.1, is abundantly expressed in neurons with the ability to fire at high frequencies [4-6]. Kv3.1 has a high activation threshold and very fast activation and inactivation rates as distinguishing features from other potassium channels [7]. Kv3.1 channels are expressed at particularly high levels in fast-firing GABAergic interneurons of the cerebral cortex and the hippocampus [6,8-10]. The localization of Kv3.1 channels in parvalbumin-expressing GABAergic interneurons suggests that an inhibition of Kv3.1 activity could cause excessive excitement of neural networks by dysfunctions of these GABAergic interneurons, resulting in chronic and repeated temporary paralysis of brain functions [11-13]. There are several evidences that epileptic seizures can be suppressed by the Kv3.1 channels. It has been reported that a recurrent de novo mutations in Kv3.1 channels cause progressive myoclonus epilepsy, an inherited disorder that induces tonic-clonic seizures and ataxia [14] and a suppression of Kv3.1 channels in cortical neurons could lead to seizure-like activity [15]. In the seizure-prone, but not in seizure-resistant, animals, the levels of neuronal Kv3.1 channel were reduced by up to 50%, suggesting that reduced levels of Kv3.1 channels in neuron can cause animals to have seizures [16]. These findings support that the Kv3.1 channel plays an importance role in setting seizure threshold.
Since rosiglitazone can inhibit voltage-gated K+ channels in vascular smooth muscle [17,18] and cloned Kv1.3, Kv1.5, and Kv4.3 channels [19-21], we hypothesize here that the use of diabetes drug rosiglitazone could inhibit Kv3.1 channels and suppress fast-firing GABAergic interneurons, and thus, elevate the overall network excitability or epileptic activity of neural networks. In this study, an experiment using the whole-cell patch-clamp technique was performed on the interaction of rosiglitazone with Kv3.1 expressed in Chinese hamster ovary (CHO) cells.
METHODS
Cell culture and transfection
The method for establishing Kv3.1 expression in CHO cells has been previously described in detail [5,22]. Briefly, rat Kv3.1b cDNA [23] was transferred into the expression vector pRc/CMV (Invitrogen Corporation, San Diego, CA, USA). CHO cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Invitrogen Corporation) supplemented with 10% fetal bovine serum, 0.1 mM hypoxanthine, and 0.01 mM thymidine. Lipofectamine reagent (Invitrogen Corporation) was used to implant the Kv3.1b expression vector into CHO cells. Transfected cells were exposed to 500 µg/ml G418 (A.G. Scientific, San Diego, CA, USA), and antibiotic-resistant cells were selected and maintained in fresh IMDM containing G418. The cultures, incubated in 95% humidified air-5% CO2 at 37°C, were passaged every 4–5 days with a brief trypsin-EDTA (Invitrogen Corporation) treatment followed by seeding onto glass coverslips (diameter: 12 mm; Fisher Scientific, Pittsburgh, PA, USA) in a Petri dish. The cells were used for electrophysiology 12–24 h after the seeding. For electrophysiological recording, cell-attached coverslips were transferred to a continually perfused (1 ml/min) recording chamber (RC-13; Warner Instrument Corporation, Hamden, CT, USA).
Electrophysiology
Kv3.1 currents were recorded in CHO cells using the whole-cell patch-clamp technique [24] at room temperature at 22–23°C. The micropipettes were fabricated from glass capillary tubing (PG10165-4; World Precision Instruments, Sarasota, FL, USA) using a two-stage vertical puller (PC-10; Narishige, Tokyo, Japan), and had a tip resistance of 2–3 MΩ when filled with a pipette solution composed of 140 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM EGTA (pH 7.3 with KOH). Whole-cell currents were amplified with Axopatch 200B amplifier (Molecular Devices, San Jose, CA, USA), digitized with Digidata 1440A (Molecular Devices) at 5 kHz and low-pass filtered with a four-pole Bessel filter at 2 kHz. Pipette and whole-cell capacitive currents were canceled by the amplifier and the series resistance was corrected to 80% with the amplifier. Leak subtraction was not used. Voltage command generation and data acquisition were controlled with pClamp 10.1 software (Molecular Devices). Recording chamber (RC-13; Warner Instrument Corporation) was continuously perfused at 1 ml/min with a bath solution composed of 140 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 20 mM HEPES and 10 mM glucose (pH 7.3 with NaOH). The vehicle for rosiglitazone (Cayman Chemical Co., Ann Arbor, MI, USA) had less than 0.1% of dimethyl sulfoxide (DMSO) and by itself, had no effect on Kv3.1 currents (data not shown).
Data analysis
Data were analyzed with Origin 7.0 (OriginLab Corp., Northampton, MA, USA) and Clampfit 10.1 software (Molecular Devices). The IC50 and Hill coefficient (n) in the dose-response curve were obtained from the following equation:
where I (%) is the percent inhibition of current (I (%) = [1 – Idrug / Icontrol] × 100) and [D] represents a variety of drug concentration. The steady-state activation curve was fitted with the Boltzmann equation:
where Imax represents the maximum current, k is the slope coefficient, V is the test potential, and V1/2 is the potential at which the conductance is half-maximal. In the second half of the activation (i.e., up from 50% to 100% of peak amplitude), the activation time was obtained by fitting it with a single exponential function. The steady-state inactivation curves were obtained using a two-pulse protocol: currents were induced by a 300 ms pulse to +40 mV while 20 sec preconditioning pulses were varied from –60 to +30 mV using 10 mV steps every 30 sec in the absence and presence of drug. The experimental points were fitted from the following equation:
in which Imax represents the current measured at the most hyperpolarized preconditioning pulse, V is the preconditioning potential, and V1/2 and k represent the potentials corresponding to the half-inactivation point and the slope coefficient, respectively. The drug-induced time constants and deactivation time constants were determined by a single exponential fit.
Results were expressed as means ± SEM. Student’s t-test and analysis of variance (ANOVA) were used for statistical analysis with a confidence level of p < 0.05.
RESULTS
Dose-dependent inhibition of Kv3.1 channel by rosiglitazone
The action of rosiglitazone on the Kv3.1 channel was investigated using a whole-cell patch-clamp technique. Whole-cell currents were elicited with 300-ms depolarizing pulses to +40 mV in CHO cells expressing Kv3.1 channel. As shown in Fig. 1A, under control conditions, Kv3.1 current rapidly was activated and then slightly inactivated during the +40 mV pulse, as previously reported [7]. Bath-applied rosiglitazone reduced the Kv3.1 currents and accelerated the rate of decay in a concentration-dependent manner (Fig. 1A). The inhibition of Kv3.1 currents for each concentration of rosiglitazone reached a stable state within 2 min. The inhibition of terminal-pulse currents by rosiglitazone was more pronounced than that of peak currents. A nonlinear least-squares fit of dose-response plots with the Hill equation yielded an IC50 value of 29.8 ± 3.3 µM and a Hill coefficient of 1.5 ± 0.2 (n = 5) for the end-pulse currents, and an IC50 value of 125.4 ± 28.8 µM and a Hill coefficient of 0.7 ± 0.1 (n = 5) for the peak currents (Fig. 1B). From this experiment, 30 µM rosiglitazone, slightly higher than the IC50 value, was used in subsequent experiments. As shown in Fig. 1C, the effect of rosiglitazone was reversible: the inhibition of end-pulse currents caused by 30 µM rosiglitazone was reversed to 88.5 ± 4.1% of the pre-drug baseline (n = 5) after 2-min wash-out.
The greater action of rosiglitazone on the end-pulse current than the peak current means that rosiglitazone can suppress Kv3.1 channels once they have been opened [21,25-32]. If rosiglitazone acts as an open channel blocker of Kv3.1, the activation time constant is expected to be unaffected by rosiglitazone. Indeed, the activation time constant of Kv3.1 currents, elicited with a 300-ms depolarizing pulse from –80 to +40 mV, was not significantly affected by rosiglitazone: 2.1 ± 0.08 ms (n = 5) under control conditions and 2.2 ± 0.07 ms (n = 5) in 30 µM rosiglitazone. Based on the difference in the rosiglitazone-mediated inhibition between end-pulse and peak currents, and no effect of rosiglitazone on the activation time constant, we hypothesized that rosiglitazone could act on the Kv3.1 current as an open channel blocker.
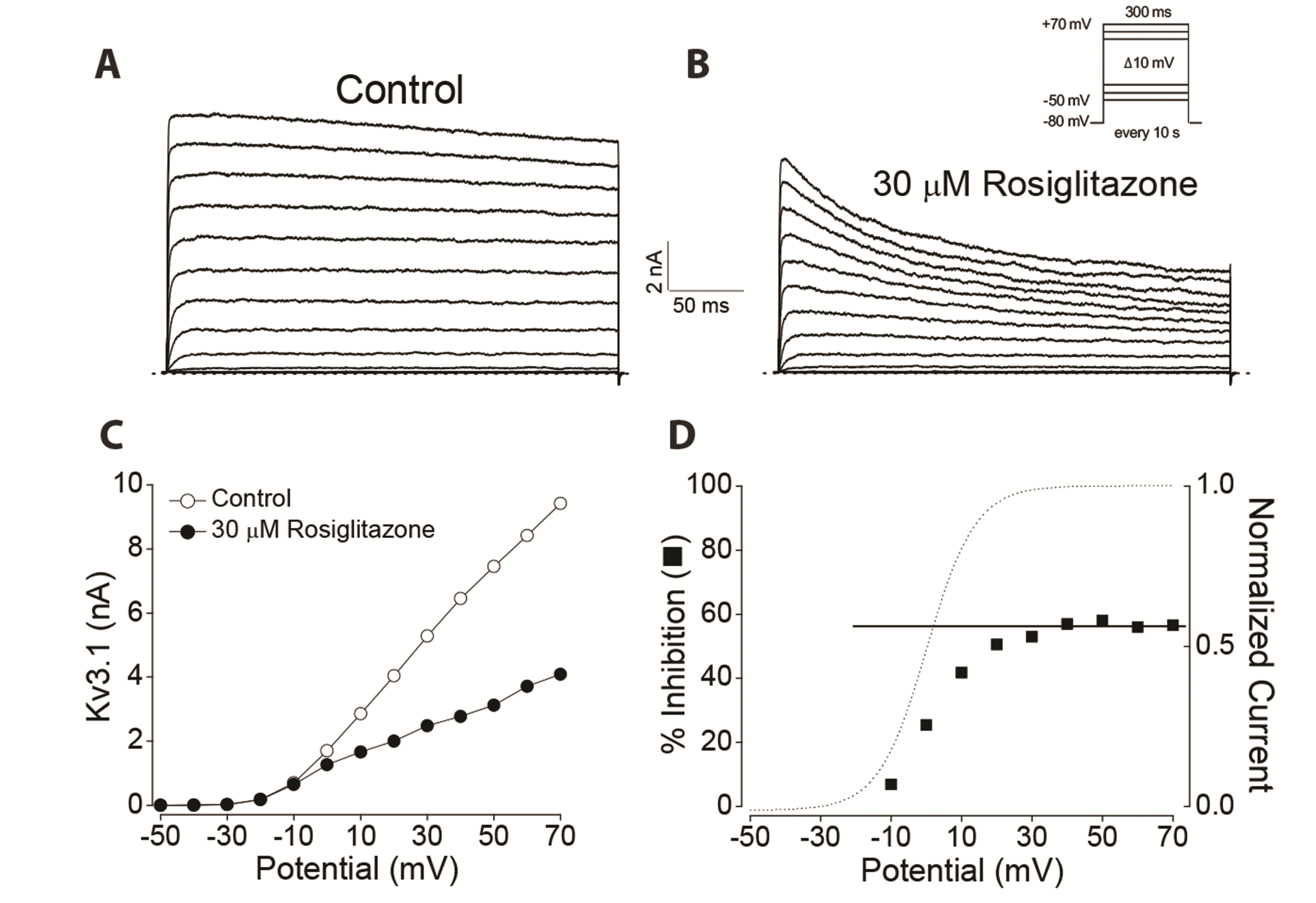
Voltage dependence of rosiglitazone-induced inhibition of Kv3.1
Next, we tested whether the inhibition of Kv3.1 currents by rosiglitazone was dependent on membrane potential. If rosiglitazone blocks the Kv3.1 channel in the open state, inhibition by rosiglitazone appears in the voltage range where the channel starts to be activated, increases in proportion to the degree of activation, and remains constant when fully activated. To examine this prediction, a current-voltage (I-V) relationship was constructed in the absence and presence of 30 µM rosiglitazone (Fig. 2A–C). In the absence of rosiglitazone, Kv3.1 currents started to be detected at –20 mV and increased with higher depolarizing pulses (Fig. 2A, C). When channel conductance was obtained from the I-V relationship, it steeply increased in a sigmoidal form in the voltage range from –20 to +30 mV and remained in a fully active state above +30 mV (Fig. 2D, dotted line). Rosiglitazone (30 µM) inhibited Kv3.1 currents over the entire voltage range in which Kv3.1 was activated, i.e., from –20 to +70 mV (Fig. 2B, C). To investigate the voltage dependence of the rosiglitazone-induced inhibition of Kv3.1 channel, percent inhibition of Kv3.1 currents (see Methods) against membrane potential was plotted (Fig. 2D). A high degree of suppression was detected with a strong voltage dependence at voltages between –10 and +30 mV, which is a gradual channel opening range (Fig. 2D). However, the suppression of Kv3.1 by rosiglitazone at potentials between +30 and +70 mV, where the channel is fully activated, did not show such voltage dependence: 54.7 ± 3.8% inhibition at +30 mV and 56.1 ± 3.4% inhibition at +70 mV (n = 4, ANOVA, p < 0.05). The linear curve fitting of the data at voltages above +40 mV (Fig. 2D, solid line) showed a slope of zero. In summary, the voltage dependent data implies that the block of Kv3.1 by rosiglitazone preferentially occurs after the channel opens. In addition, the voltage-independent effect of Kv3.1 currents by rosiglitazone indicates that the effect of rosiglitazone is independent of membrane potential once the channel is fully activated.
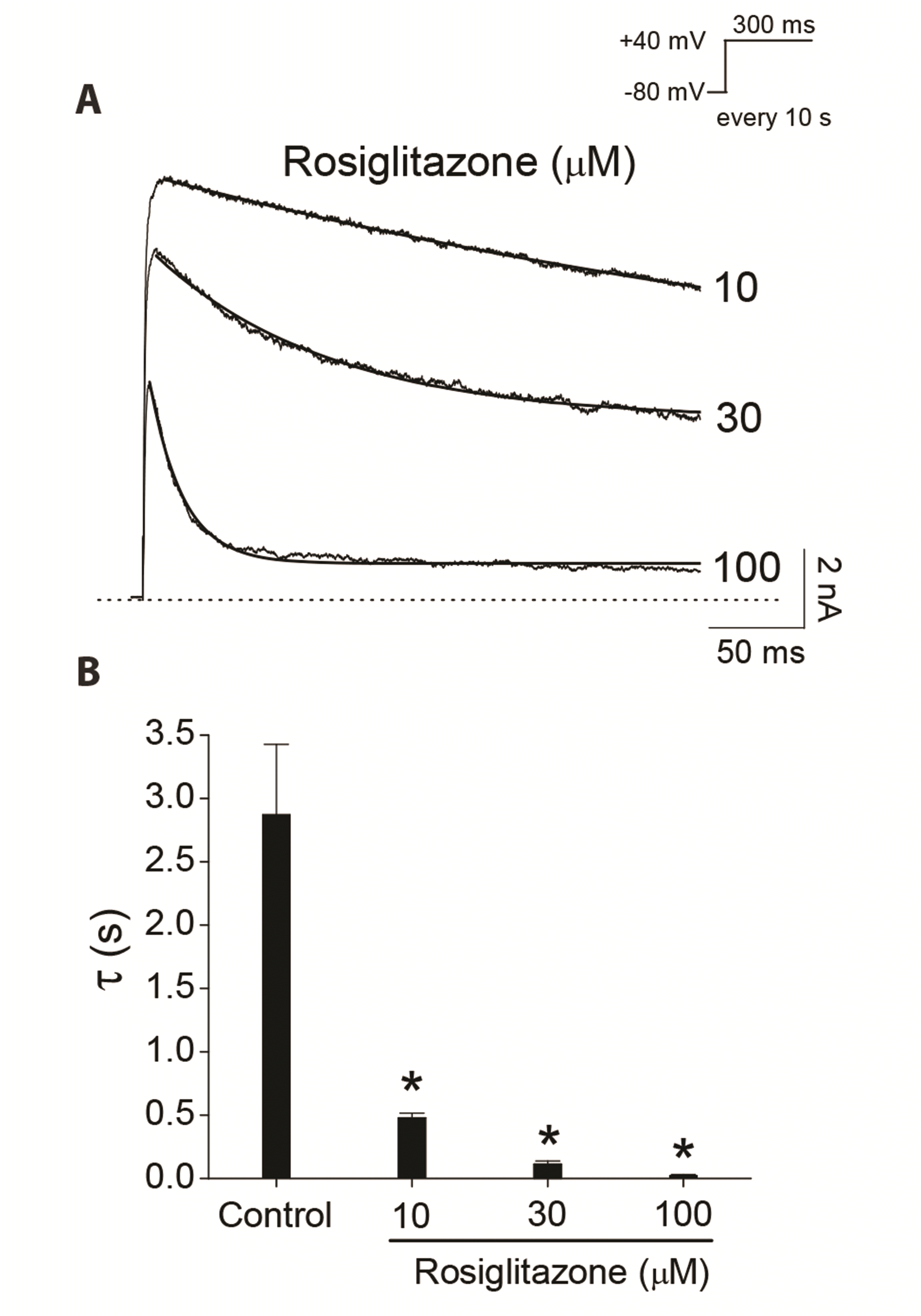
Kinetics of Kv3.1 current decay and deactivation by rosiglitazone
Fig. 3A shows that rosiglitazone accelerated the decay of Kv3.1 current during the 300-ms depolarizing pulse at +40 mV in a concentration-dependent manner. Time constants were obtained with a single exponential fit to the traces of current decay at 10, 30, 100 µM of rosiglitazone. The time constant value obtained at low concentration (3 μM) of rosiglitazone was excluded to prevent contamination by the time constant of the intrinsic slow inactivation of the Kv3.1 currents under control conditions. Fig. 3B shows a summary of the time constants at various concentrations of rosiglitazone. The time constant value under control conditions was 2.9 ± 0.6 sec (n = 4). The time constant values decreased as the concentration of rosiglitazone increased, suggesting that the concentration-dependent rosiglitazone-induced current decay of Kv3.1 is associated with time-dependent development of the block by the drug once Kv3.1 is opened.
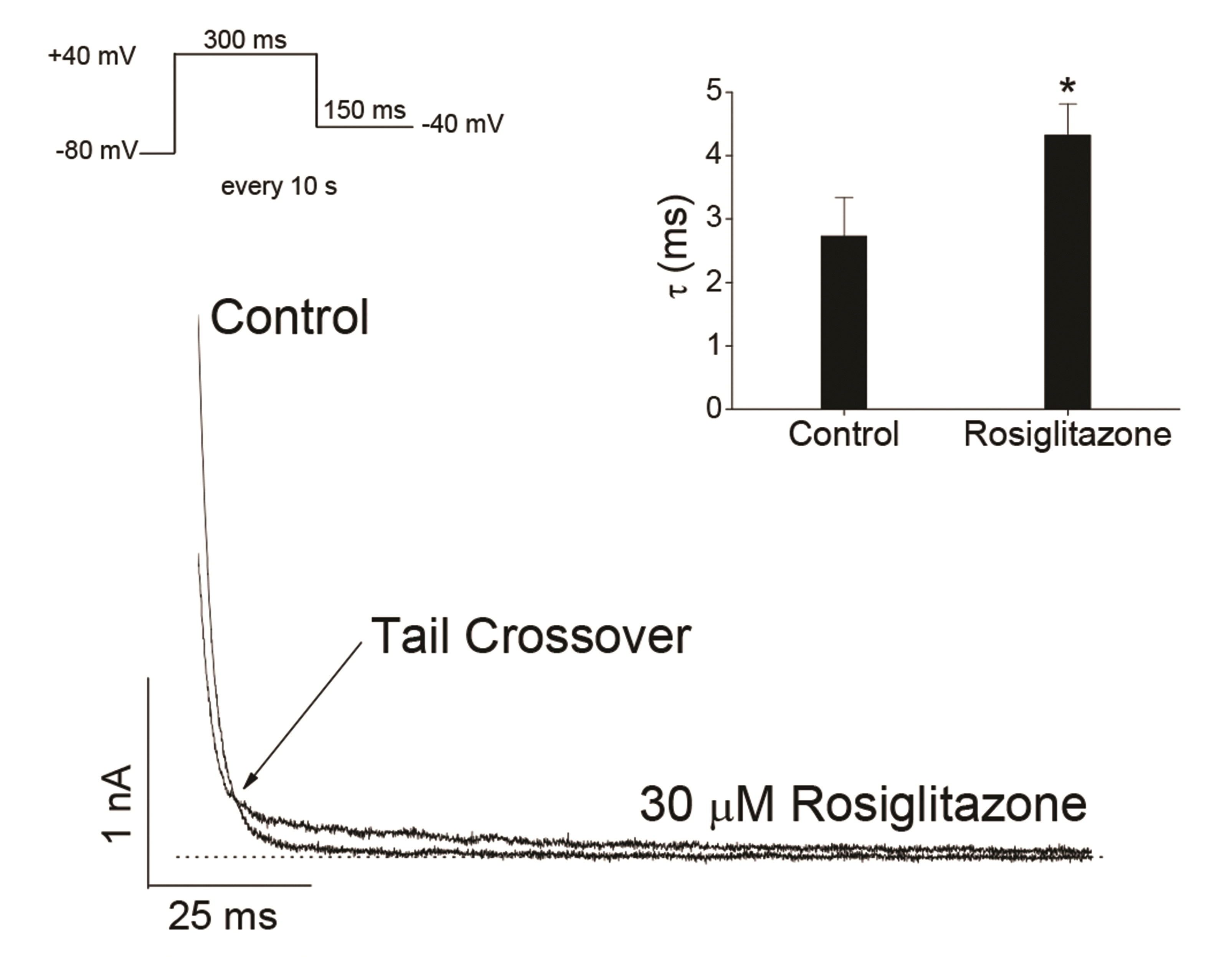
Information on open channel blockers can be obtained through their action on deactivation kinetics. If a blocker binds to channel in its open state, the closing rate of the channel may be slower compared to the closing rate of the channel without the drug [19,21,25,29,33]. The effect of rosiglitazone on Kv3.1 current deactivation kinetics was investigated using tail currents of Kv3.1 channel. Fig. 4 shows superimposed representative tail currents, which were activated by returning the 150-ms repolarizing pulse of –40 mV after Kv3.1 activation by a 300-ms depolarizing pulse of +40 mV from a holding potential of –80 mV, under control conditions and in the presence of 30 µM rosiglitazone. The control tail currents declined quickly with a time constant of 2.7 ± 0.6 ms (n = 4) when fitted with a single exponential function. In the presence of 30 µM rosiglitazone, the initial amplitude of tail current was markedly decreased and the subsequent decay time course of tail currents was slowed with a time constant of 4.3 ± 0.5 ms (n = 4) when fitted with a single exponential function (p < 0.05). Because rosiglitazone decreased the initial amplitude of the tail current and slowed down the decay kinetics, superimposing the two tail currents in the absence and presence of rosiglitazone showed a "tail crossing" phenomenon (Fig. 4).
Use-dependent inhibition of Kv3.1 by rosiglitazone
Open channel blockers generally exhibit use-dependent inhibition because the more frequently the channel is opened, the more likely the blocker is to bind to the channel pores. If rosiglitazone blocks the open pore of the Kv3.1 channel as shown in the analysis results (Figs. 2–4), rosiglitazone is predicted to block the channel in a use-dependent manner. To examine this prediction, we repetitively evoked Kv3.1 currents with a 300-ms depolarizing pulse of +40 mV from a holding potential of –80 mV, 8 times at 1 Hz and 16 times at 2 Hz, respectively. For the two different stimuli, the duration of stimuli is equal to 8 sec. After 2 min exposure to 30 µM rosiglitazone at –80 mV without depolarization, the 1 Hz and 2 Hz depolarization protocols were resumed. Without rosiglitazone, the progressive inhibition of Kv3.1 current was negligible during the 1 Hz stimuli (Fig. 5A). The peak amplitude of the 8th Kv3.1 current was slightly decreased by 9.3 ± 1.9% (n = 4) compared to the amplitude of the first current. However, in the presence of 30 µM rosiglitazone, the peak amplitudes of Kv3.1 currents were progressively decreased by a significantly larger magnitude, 17.0 ± 3.4% (n = 4; p < 0.05), at the end of the 1 Hz train (Fig. 5A, B). This effect of rosiglitazone was also observed when Kv3.1 was activated at 2 Hz. At the end of the 2 Hz train, the peak amplitudes of the Kv3.1 current in the absence and presence of 30 µM rosiglitazone were decreased by 24.3 ± 3.4% (n = 4) and 35.8 ± 4.9 (n = 4), respectively (p < 0.05; Fig. 5B). In the presence of rosiglitazone, the Kv3.1 currents showed a robust use-dependent reduction at 1 Hz and 2 Hz compared to the non-rosiglitazone control group. The results of greater use dependence with rosiglitazone suggests that rosiglitazone might block the open channel of Kv3.1. It is interesting to note that without activation of the Kv3.1 channel, 30 µM rosiglitazone itself had little effect on the current amplitude, as the first current in the stimulus set did not differ significantly between the control and rosiglitazone conditions. This also strongly indicates that rosiglitazone may not bind to the closed Kv3.1 channel.
If the block of Kv3.1 by rosiglitazone is use-dependent, the magnitude of the blockade will increase with the degree of use or channel opening. The use-dependent inhibition with 2 Hz stimuli was appeared to be more pronounced than that with 1 Hz activation in rosiglitazone (Fig. 5B). However, the gradual reduction of the control current by repeated stimulation obscured the correct determination of the comparison between 1 Hz and 2 Hz stimulation. To eliminate this problem, we normalized the currents in rosiglitazone to the corresponding control currents in a series of stimuli (Fig. 5C). Plots of normalized currents versus the elapsed time showed a greater blockade at 2 Hz, further confirming the use dependence of rosiglitazone, and these results are also in line with the idea that rosiglitazone can block open channels of Kv3.1.
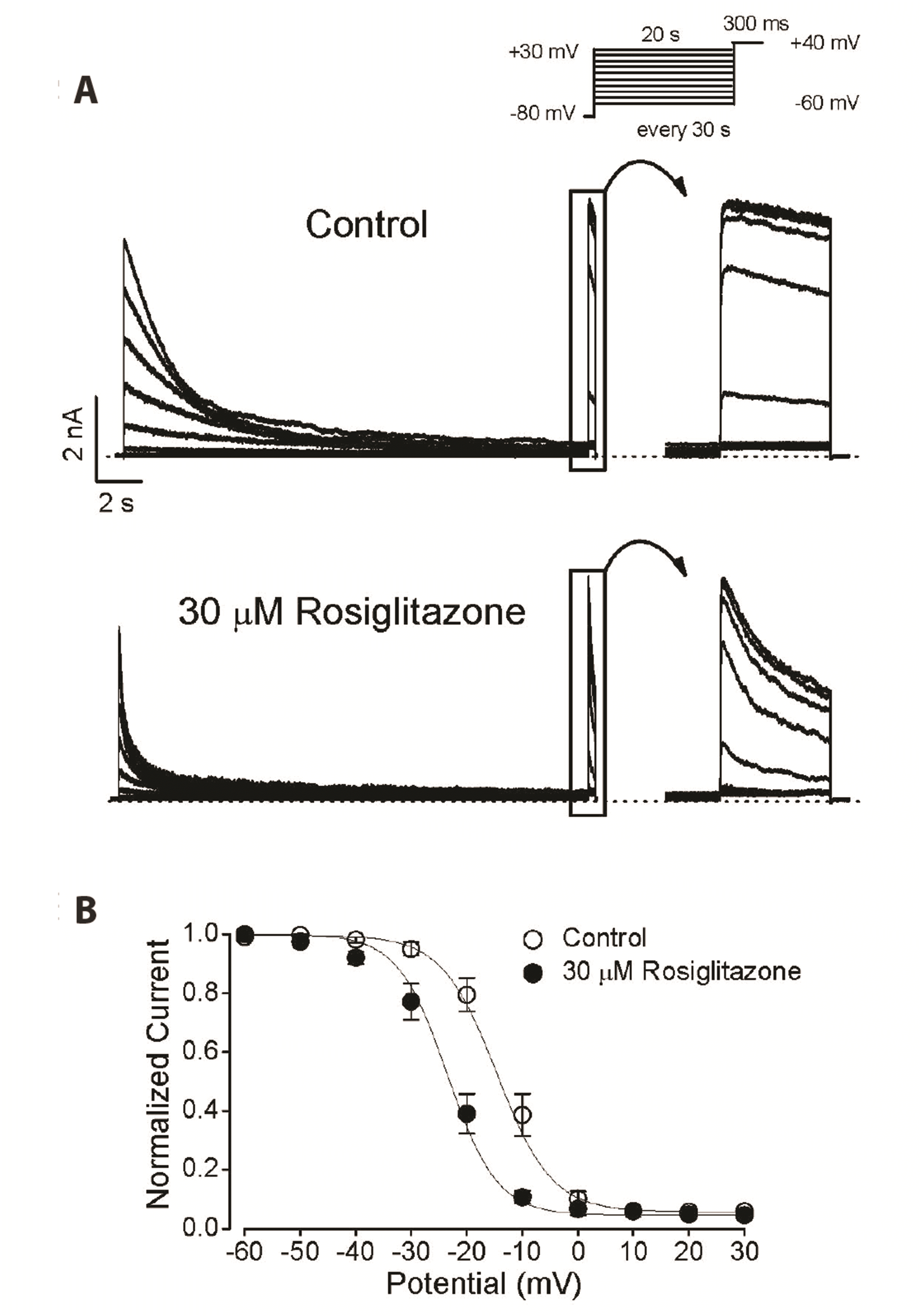
Effects of rosiglitazone on steady-state inactivation of Kv3.1
The Kv3.1 current is rapidly activated at the depolarizing membrane potential and then slightly inactivated during the pulse [7]. Based on the results of this study, rosiglitazone decreased the Kv3.1 current and accelerated the decay rate, which can be interpreted as acting as an open channel blocker. However, the acceleration of current decay in the presence of rosiglitazone might be due to drug-induced acceleration of the intrinsic slow inactivation process of the channel, followed by transition of the open state to the inactive state. We here examined the effect of rosiglitazone on the steady-state inactivation of Kv3.1 channel. Fig. 6A shows the steady-state inactivation current traces of Kv3.1 in the absence and presence of 30 µM rosiglitazone obtained using a typical two-pulse protocol (see Methods). As shown in Fig. 6B, in the control condition, the potential for the half inactivation point (V1/2) and slope coefficient (k) of the steady-state inactivation curve were −14.6 ± 1.1 and 5.0 ± 0.4 mV, respectively (n = 4). In the presence of rosiglitazone (30 µM), the potential for the V1/2 and k were −23.5 ± 2.3 and 4.8 ± 0.5 mV, respectively (n = 4). Rosiglitazone significantly shifted the inactivation curve to a negative potential (p < 0.05) without a significant change in the k value. Based on these results, the inhibition of Kv3.1 current by rosiglitazone might be due to the acceleration of the intrinsic and slow inactivation process of the channel once the drug binds to the inactivated state of the channel. This aspect will be discussed further.
DISCUSSION
This study shows that the antidiabetic drug rosiglitazone blocks the Kv3.1 channel expressed in CHO cells. Inhibition of Kv3.1 by rosiglitazone is achieved through a concentration-dependent acceleration of the apparent rate of current decay. These results are very similar to various open channel blockers[21,25-27,29,30,32-36]. The characterization of Kv3.1 inhibition by rosiglitazone suggests that rosiglitazone preferentially interacts with the open state of Kv3.1 channels based on the following evidence. 1) Rosiglitazone accelerated the decay rate of Kv3.1 current during the depolarization pulse (Figs. 1, 3). 2) At the onset of the depolarization pulse, rosiglitazone did not affect the initial time course of channel activation. The data that the activation time constant was not affected by rosiglitazone as illustrated in the results (Fig. 1) indicates that rosiglitazone does not bind to the closed or resting state of Kv3.1. 3) The inhibition induced by rosiglitazone was increased steeply in the voltage range of channel activation (Fig. 2). 4) Rosiglitazone slowed the deactivation time course, resulting in tail crossover (Fig. 4). This phenomenon suggests an interaction between rosiglitazone and the open state of Kv3.1 [19,21,25-27,29,32,33]. 5) The suppression of Kv3.1 channels by rosiglitazone is use-dependent and is enhanced by a higher rate of channel activation (Fig. 5). This is consistent with the action of rosiglitazone on open-state Kv3.1 [21,25-27,29,32]. 6) Rosiglitazone did not inhibit the Kv3.1 current without activation of the channel as illustrated in the results (Fig. 5). These results strongly suggest that the drug does not interact with Kv3.1 in the closed or resting state.
Drugs that interact primarily with the open state of the channel can do so by migrating into the ion-conducting pores. A high level of inhibition was detected with a strong voltage dependence at voltages in the gradual channel opening range, whereas suppression of Kv3.1 by rosiglitazone was voltage-independent at the voltage at which the channel was fully activated. As the pKa values of rosiglitazone are 6.1 and 6.8 [37], it is mainly uncharged at intracellular or extracellular pH 7.3 (pH of the pipette or bath solution). Therefore, no additional block was detected in the voltage range where the channels are fully activated (Fig. 2). In the voltage range in which the channels are fully activated, the degree of probability of ion channel opening does not change any more even if the membrane depolarization increases. These results suggest that the interaction between rosiglitazone and Kv3.1 is independent of transmembrane.
In this study, another feature of the rosiglitazone-mediated inhibition of Kv3.1 is that the steady-state inactivation curve of Kv3.1 shifts to a negative potential without a significant change in the slope value. This result suggests that inhibition of Kv3.1 current by rosiglitazone may be due to the acceleration of the channel's intrinsic slow inactivation process once the drug binds to the inactive state of the channel. Similar previous results show that open channel blockers shifted steady-state inactivation of ion channels [38-41]. In addition, use-dependent inhibition can be achieved by binding of the drug to an open and/or inactivated states of the channels [29,38,40,42], indicating that an open channel blocker may have secondary access the binding site in the inactive state of the channel at a voltage range lower than the activation threshold. Another interpretation is that once bound to an open channel, rosiglitazone can become trapped in the channel, and consequent conformational changes in the channel can accelerate the inactivation process and prolong the dwelling time in the inactivated state. Further elucidation of the mechanism will be interesting in future studies.
Based on the pharmacokinetics of orally administered rosiglitazone to patients with type 2 diabetes, therapeutic human plasma concentrations of rosiglitazone have been reported to differ between 0.3–2 μM in patients with diabetes [2,43,44]. Although this plasma level is lower than the IC50 value of rosiglitazone in this study, the drug concentration in the tissue may be higher than in plasma because of its high lipophilicity and affinity for adipose tissue. All these unique properties of cells in vivo may limit direct extrapolation of our data to clinical settings. Nevertheless, our study clearly shows that rosiglitazone acts as an open channel blocker of the Kv3.1 channel.
Kv3.1 channels are known to express at particularly high levels in fast-firing GABAergic interneurons expressing parvalbumin of the cerebral cortex and the hippocampus [6,8-10], suggesting that blockade of Kv3.1 channels could reduce interneuronal spiking, resulting in weakened GABAergic inhibition and hence an increase in the net excitability of neural networks[14-16]. Because our data indicate that the blockade of Kv3.1 channels by rosiglitazone is use-dependent, the suppressive effect of rosiglitazone, if any, on fast spiking of interneurons could become more pronounced during repetitive firing. The very similar biophysical properties between cloned Kv3.1 channels and the neuronal endogenous Kv3.1 channels [4-6,45] suggests that the rosiglitazon effects observed in this study could be replicated in intact neurons, but this issue has not yet been tested. If rosiglitazone possesses pro-convulsive effects, extra caution is required when administering it to diabetic patients. Future studies should focus on the detailed characterization of the extent and strength of pro-convulsive activity of rosiglitazone.
In conclusion, this study demonstrated the inhibitory effects of rosiglitazone on the Kv3.1 expressed in CHO cells for the first time. A detailed study of the kinetics of rosiglitazone action on Kv3.1 suggests that rosiglitazone is an open-channel blocker for Kv3.1 in a concentration-, voltage-, time-, and use-dependent manner, implying that rosiglitazone might suppress fast-firing GABAergic interneurons, and thus, elevate the overall network excitability or epileptic activity of neural networks.
 XML Download
XML Download