

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Osteoporosis is a systemic skeletal disorder that causes vulnerability of bones to fracture due to reduction in bone density and degradation of the microstructure of bone tissues [1]. Osteoporosis is the most common metabolic bone disease and affects half of female and one-third of male in their sixties and seventies. Prevention and treatment of osteoporosis are critical because the increased prevalence of osteoporosis and fragility fractures, along with aging of the global population, result in significant clinical, economic, and social burdens [2-4].
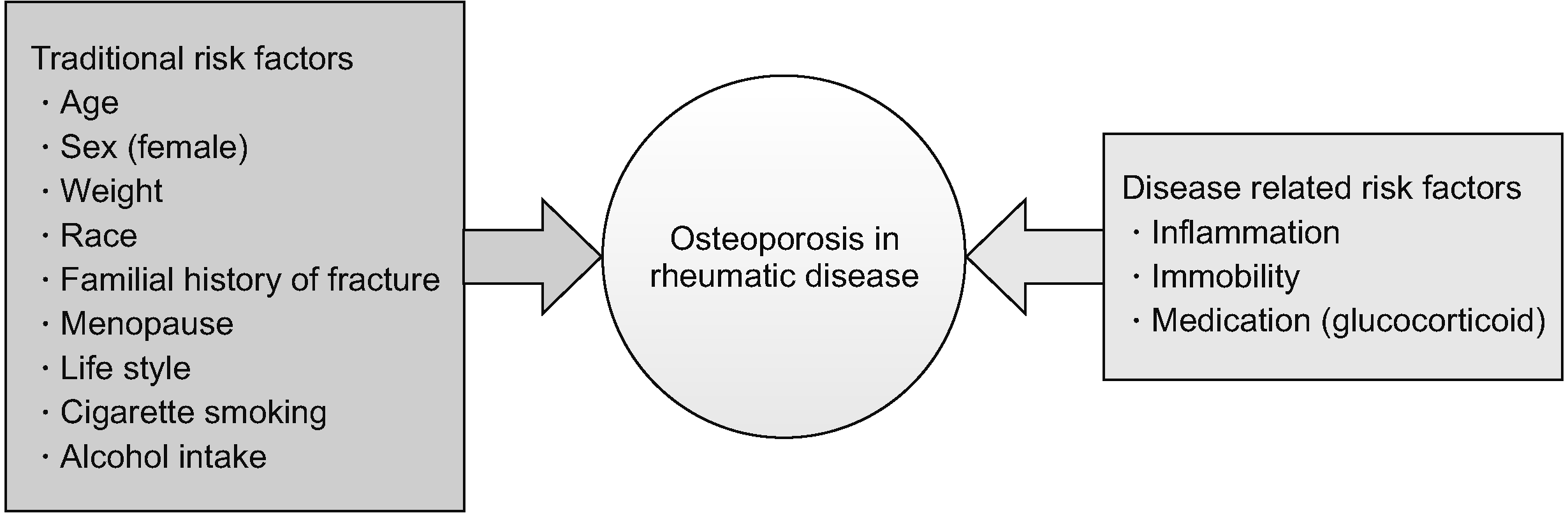
Bones in the human body maintain a healthy state through a remodeling process. Bone remodeling is a physiological process of maintaining the optimal bone state by removing aged and damaged bone cells and generating new ones. In this process, the receptor activator of NF-κB (RANK), receptor activator of NF-κB ligand (RANKL), and osteoprotegerin (OPG) pathways are important for osteoclastogenesis. In addition, the Wnt signaling pathway has an important role in osteoblastogenesis. The above pathways as well as immune cells and cytokines are linked with bone cells and are regulated precisely. Osteoporosis is induced by an imbalance between bone formation and bone resorption in the bone remodeling process. The risk of osteoporosis and fragility fractures is higher in patients having autoimmune rheumatic diseases than in the general population. In autoimmune rheumatoid diseases, in addition to the traditional risk factors, various factors such as chronic inflammation, autoantibodies, decreased physical activity due to arthritis, drugs, and vitamin D deficiency can contribute to bone loss (Figure 1) [5,6].
Rheumatoid arthritis (RA) is a common systemic autoimmune disease with an unknown cause and is characterized by chronic, symmetric, and progressive inflammatory polyarthritis, in which bone loss can be caused by chronic inflammation [7,8]. Osteoporosis is more common in RA than in the general population. The prevalence rates of osteoporosis and osteoporotic fracture in both males and females are 2 and 1.5 times higher than those of the general population, respectively [9,10]. In RA patients, an increase in fracture frequency was observed in patients with higher bone mineral density (BMD) than in the general population, suggesting bone quality deterioration in RA patients [11]. Pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, IL-17, and tumor necrosis factor (TNF)-α, which play important roles in the pathogenesis of RA, not only induce chronic inflammation, but also promote bone loss and fragility through increased RANKL expression. They have been found to foster bone loss through processes such as resorption imbalance. These findings suggest that modulating inflammation in RA may be important in suppressing bone loss [12-14]. In support of this, biologic disease-modifying anti-rheumatic drugs (bDMARD) and targeted synthetic DMARDs (tsDMARDs), which have been widely used for treatment of RA, not only modulate disease activity, but also inhibit local and systemic bone loss [15].
In this review, we summarize the effects of inflammatory and autoimmune responses that play important roles in RA pathogenesis, and we identify the key pathways, cells, and mediators involved in bone resorption and bone formation in osteoporosis. Since relatively few studies have investigated the effects of tsDMARD on bone, here we discuss whether biologics have a positive effect on osteoporosis in RA.
MAIN SUBJECTS
Pathogenesis of osteoporosis in RA
1) Cells and osteoporosis
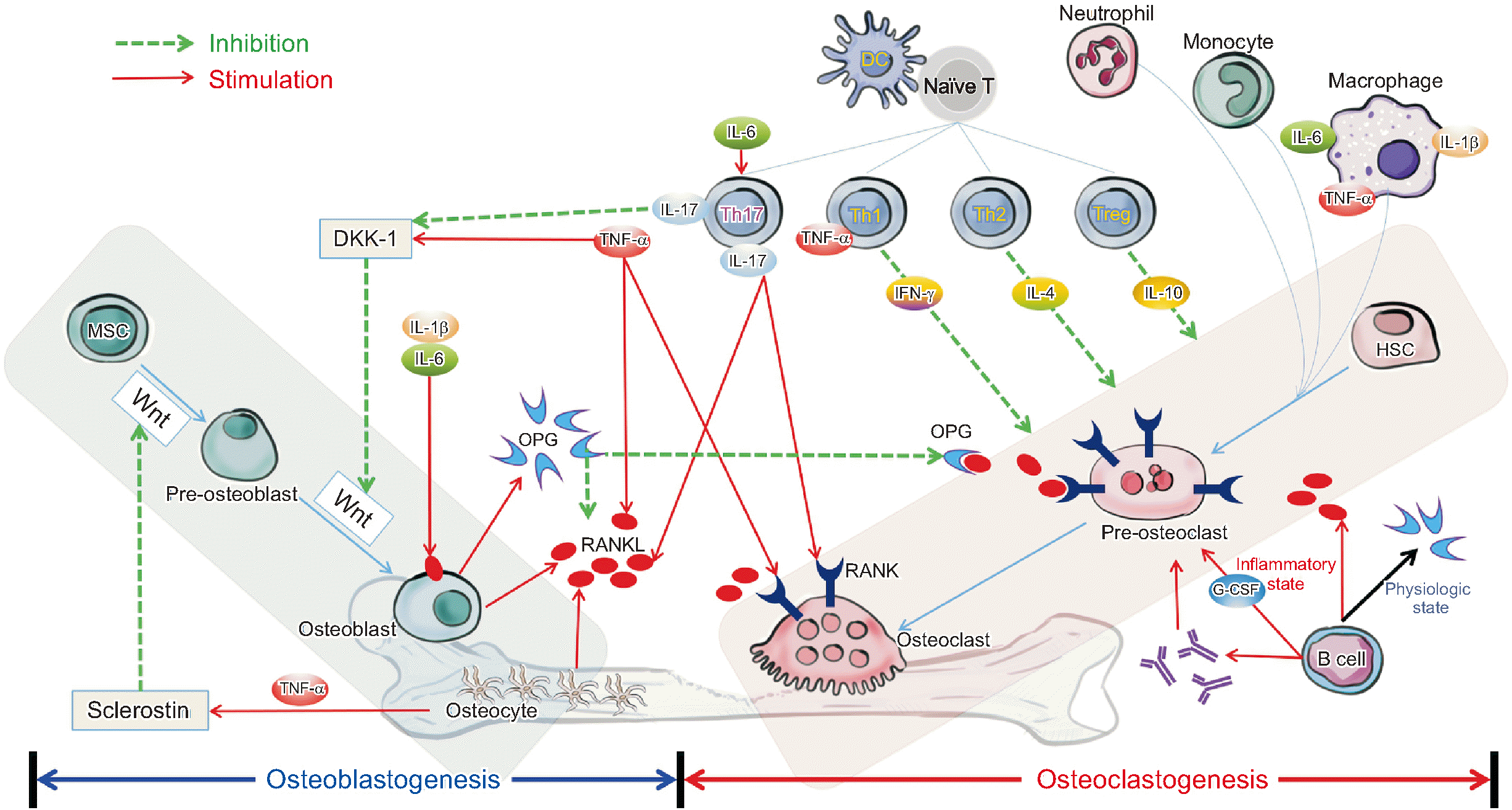
Bone is a dynamic organ that continually changes and undergoes continuous remodeling throughout life. Several factors are involved in bone remodeling, among which the interactions between bone cells (osteoclasts, osteoblasts, and osteocytes) and immune cells (macrophages, monocytes, dendritic cells, T cells, and B cells) are very important (Table 1, Figure 2) [16-21].
(1) Bone cells (osteoclast–osteoblast–osteocyte)
The osteoclast is a multinuclear cell in the monocyte/macrophage line derived from the hematopoietic stem cell. It has an important function in maintenance and remodeling by decomposing organic and inorganic parts of bone tissue. In the presence of inflammation, the influx of osteoclast precursors and differentiation and activation into osteoclasts can be enhanced by pro-inflammatory cytokines, such as macrophage colony-stimulating factor (M-CSF), IL-1β, IL-6, IL-17, and TNF-α [22]. The RANKL-RANK-OPG system plays a critical role in osteoclast regulation.
Osteoblasts, which are derived from mesenchymal stem cells, are responsible for bone synthesis and mineralization through the processes of bone modeling and remodeling. In addition, these cells produce various cell products, such as alkaline phosphatase (ALP) and collagenase, growth factors, osteocalcin, and collagen. The Wnt pathway is crucial to the differentiation and function of osteoblasts [23].
Osteoclast activity is regulated by the interaction of OPG and RANKL produced by osteoblasts [24]. RANKL is a key molecule involved in the regulation of osteoclast differentiation and is mainly produced by osteoblasts and osteocytes. In addition, RANKL can be generated from synovial fibroblasts and activated B cells and T cells (especially Th17 cells) under inflammation [25,26]. RANKL binds to RANK in osteoclast precursors and mature osteoclast to induce differentiation and activation. This process can be inhibited by OPG, which acts as a soluble decoy receptor for RANKL. Pro-inflammatory cytokines, such as TNF-α, which are increased in inflammatory rheumatic diseases, inhibit Wnt-induced osteoblastogenesis through increased expression of dickkopf-1 (DKK1), a Wnt pathway inhibitor [27-29], while promoting osteoclastogenesis by increasing RANKL expression.
Osteocytes compose 90%~95% of all bone cells of the adult skeleton, and their average half-life is approximately 25 years [30]. Osteocytes are derived from mesenchymal osteoprogenitor cells and osteoblasts. They are located in a space called the lacunae within a mineralized matrix. Osteocytes form networks with tiny canals called caniculi. Through this network, nutrients, excreta, and other materials are exchanged. In addition, it plays important roles as a sensor of mechanical load, regulator of bone remodeling through control of phosphate homeostasis and endocrine cell secretion of various factors, and regulator of osteoblast and osteoclast activity [31].
(2) T cells
The naive cluster of differentiation 4+ (CD4+) T cell is differentiated into Th1 (T helper 1), Th2, Th9, Th17, Th22, Treg (regulator T), and Tfh (follicular helper T) cells by various micro-environmental and cytokine stimuli. Each performs its own function [32]. The T cells that are critical in RA pathogenesis are Th1, Th2, Th17, and Treg. Their effects are outlined below.
Th1 cells: Naive CD4+ T cells differentiate into Th1 cells when stimulated by IL-12 to produce interferon gamma (IFN-γ), IL-2, lymphotoxin, TNF-α, and granulocyte-macrophage CSF (GM-CSF) [33]. Through this process, they generally induce cell-mediated immune responses by macrophage and cytotoxic T cells against intracellular bacteria and protozoa [34]. Th1 cells have also been implicated in the pathogenesis of various autoimmune and inflammatory diseases [34-36]. In bone metabolism, Th1 cells may inhibit osteoclastogenesis. IFN-γ, a Th1 cytokine, can reduce bone loss by inhibiting osteoclast formation through degradation of TNF Receptor Associated Factor 6 (TRAF6) molecules.
Th2 cells: The Th2 cells induce humoral immune responses and play an important role in host defense against parasites and allergic and atopic diseases [37]. These cells mainly regulate eosinophils, basophils, and mast cells. However, overactivation of Th2 cells can cause diseases such as type I hypersensitivity, allergic rhinitis, atopic dermatitis, and asthma [38]. IL-4 and IL-13, which are important cytokines of Th2 cells, are associated with inhibition of osteoclastogenesis via decreased RANKL and RANK and increased OPG [39]. Th2 cells maintain osteoblast function under various inflammatory conditions through increased parathyroid hormone (PTH) production and inhibit bone loss by decreasing the RANKL/OPG ratio [40]. Therefore, Th2 cells have an osteoprotective function in the pathophysiology of osteoporosis.
Th17 cells: When naive CD4+ T cells are stimulated by IL-1β, IL-6, IL-23, and transforming growth factor-β (TGF-β), they differentiate into the Th17 cells IL-17, IL-21, IL-22. GM-CSF is the main effector cytokine [41,42]. Th17 cells protect the body from bacterial infection and induce various immune responses through generation and recruitment of neutrophils [43,44]. In addition, Th17 cells play an important role in various inflammatory diseases, including osteoporosis, psoriasis, periodontal disease, RA, and inflammatory bowel diseases [45,46]. Th17 cells are involved in osteoclastogenesis through various mechanisms. Secretion of IL-17 promotes RANKL production in fibroblasts and osteoblasts, stimulates M-CSF and RANKL production by osteoblasts and stromal cells, and increases RANK expression in osteoclast precursors [47,48]. Although IL-17 appears to have a key function in the pathogenesis of RA, treatment with IL-17 inhibitors does not achieve the primary endpoint of American College of Rheumatology 20 (ACR20), unlike in psoriasis, psoriatic arthritis, and ankylosing spondylitis [48]. In this regard, further research is needed.
Treg cells: Treg cells (CD4+CD25+Foxp3+ T cells) play an important role in controlling and preventing various inflammatory and autoimmune diseases by inhibiting activation of the immune system [49]. Treg cells regulate inflammation and immune responses by secreting anti-inflammatory cytokines, such as IL-10 and TGF-β [50,51]. They directly inhibit osteoclasts by suppression of RANKL and M-CSF production [4]. They are also known to promote activation of osteoblasts [52,53].
In summary, it is assumed that the balance among Th1, Th2, and Treg cells, which suppress osteoclastogenesis, and Th17 cells, which promote osteoclastogenesis, is critical in the pathogenesis of osteoporosis. Meanwhile, in inflammatory conditions such as RA, depending on the environment, Th1 cells can act differently.
(3) B cells
The B lymphocyte is responsible for humoral immunity among adaptive immunities. When naive B cells are activated by antigens, they differentiate into plasma cells and produce antibodies to show an immune response. Under physiological conditions, B cells produce approximately 40%~60% of OPG [54]. However, B cells activated by inflammation increase osteoclast precursors through granulocyte CSF (G-CSF) secretion [55], and they activate osteoclast formation by producing RANKL. In addition, B cells can stimulate osteoclasts through production of immune complexes and autoantibodies [56-58]. In conclusion, activated B cells in inflammatory conditions contribute to osteoclastogenesis by secreting G-CSF and RANKL while contributing to antibody production.
(4) Innate immune cells
Macrophages are inflammatory cells that contribute to the host defense system by inducing phagocytosis and inflammatory responses against pathogen infection. Macrophages act as precursors to osteoclasts and induce bone loss by producing pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α to promote osteoclast formation [59]. They also play an important role in the pathogenesis of RA [60].
Monocytes migrate from blood vessels to inflammatory sites in a C-C chemokine receptor type 2 (CCR2)-dependent manner and differentiate into macrophages or dendritic cells, contributing to inflammatory and repair processes [61,62]. Meanwhile, monocytes act as osteoclast precursors and can be involved in inflammatory cell recruitment and osteoclastogenesis through production of cytokines and chemokines [63,64].
Neutrophils are involved in the pathogenesis of various diseases, including inflammation-mediated bone loss, through cytokine and chemokine production [65-67]. Neutrophils can also differentiate into osteoclast precursors when activated. In addition, neutrophils express membrane-bound RANKL (mRANKL) and RANK and may contribute to osteoclastogenesis by acting on osteoclasts and their precursors [67,68].
Several other innate immune cells may also be directly/indirectly involved in osteoclastogenesis [20].
In summary, some innate immune cells can be differentiated into osteoclasts by serving as osteoclast precursors in the microenvironment of RANKL, M-CSF, and pro-inflammatory cytokines. These cells can also contribute to osteoclastogenesis by directly producing pro-inflammatory cytokines.
2) Cytokines and osteoporosis
TNF-α plays an important role in the pathogenesis of osteoporosis as well as in inflammatory diseases such as RA [69-72]. TNF-α increases RANKL expression in T cells, B cells, and osteoblasts, and it increases RANK expression in osteoclast precursors. Eventually, it promotes differentiation into osteoclasts and causes bone resorption [73]. Meanwhile, TNF-α inhibits osteoblast differentiation by increasing DKK1 and sclerostin expression to reduce bone formation [74,75]. As a result, TNF-α exhibits an overall osteoclastogenic effect.
IFN-γ exhibits a dual effect on bone. It can inhibit osteoclast formation by inducing degradation of the RANK adapter protein TRAF6, delaying osteoclast formation by acting directly on osteoclast precursors, and promoting osteoclast apoptosis by inducing superoxide production. On the other hand, IFN-γ acts as an inducer of major histocompatibility complex (MHC) class II expression and antigen presentation, and it can promote osteoclastogenesis by activating T cells and promoting RANKL and TNF-α secretion from T cells. It contributes to the formation of functional multinuclear osteoclasts by fusion of osteoclasts [76]. The mechanism of these conflicting reactions is not clear; nevertheless, IFN-γ is presumed to exhibit pro- or anti-resorptive activity depending on the state of estrogen or inflammation in the body.
IL-1β promotes osteoclastogenesis by increasing RANKL production in osteoblasts [77], and it inhibits osteoclast apoptosis through M-CSF production [78]. It also promotes RANKL-dependent osteoclast differentiation through activation of NF-κB and AP-1, while contributing to osteoclast migration and activation through increased C-C chemokine receptor type 7 (CCR7) [79]. In addition, it is possible to reduce osteoblast activity by inhibiting the production of ALP [77].
IL-6 can directly stimulate bone loss in RA and promotes bone resorption through increased expression of RANKL in osteoblasts, fibroblasts, and T cells. In addition, it can indirectly contribute to bone loss by stimulating the differentiation of Th17 cells [80,81]. IL-6 plays an important mediator role in the inhibition of osteoblast differentiation by TNF-α, inhibiting the Wnt pathway [82].
IL-17 is involved in RANKL-RANK-OPG pathway-dependent osteoclastogenesis. By increasing RANK expression in osteoclast precursors, it increases the sensitivity of osteoclast precursors to RANKL [83]. This encourages osteoclastogenesis by increasing RANKL expression in various cells, such as osteoblasts, synovial cells, and mesenchymal cells, and by increasing the RANKL/OPG ratio [84]. IL-17 also indirectly contributes to bone loss by inducing the expression and secretion of pro-inflammatory cytokines such as IL-6, G-CSF, GM-CSF, and TNF-α from other cells [85,86]. It has been reported that IL-17, unlike TNF-α, can promote the Wnt pathway by inhibiting DKK1 [87,88]. This may explain some of the differences between the TNF inhibitor and IL-17 inhibitor for new bone formation in spondyloarthritis.
Under the inflammatory state of RA, pro-inflammatory cytokines such as IL-1β, IL-6, IL-17, and TNF-α are increased. An increase of pro-inflammatory cytokines induces promotion of osteoclastogenesis and inhibition of osteoblastogenesis through interaction with immune cells and bone cells, contributing to local and systemic bone loss in RA (Table 2, Figure 2) [25,89].
3) Autoantibodies and osteoporosis
Autoantibodies such as rheumatoid factor (RF) or anti-citrullinated protein antibodies (ACPA) in RA are related to extra-articular involvement and poor prognosis. Their importance in osteoporosis has been highlighted in several studies. In an experimental study, citrullinated vimentin, which is a target antigen of ACPA, was expressed in monocyte/macrophage line cells and osteoclast precursors. When ACPA was bound, differentiation into osteoclast occurred [90]. In addition, IL-8 production is increased in ACPA-stimulated osteoclasts, and circulating ACPA promotes osteoclastogenesis through an IL-8-dependent autocrine loop [91]. ACPA can directly/indirectly activate osteoclasts through several mechanisms such as stimulation of TNF-α production and synovial fibroblast migration [91,92].
Autoantibody-positive cases in asymptomatic healthy individuals increase the future risk of RA in these individuals [90,93]. In a preclinical stage, an ACPA-positive group had decreased cortical bone thickness and increased porosity compared to the negative control group, as assessed by high-resolution peripheral quantitative computed tomography [94].
In addition, in a study of 155 treatment-naive patients with early-stage RA, decreased systemic BMD was observed in association with ACPA positivity and high RF level [95]. These results suggest that autoantibodies can act directly on bone. Moreover, seropositive RA patients have more severe bone erosion and osteopenia than seronegative RA patients [96-100]. A recent study also found that ACPA-positive RA patients had a higher 10-year risk of major hip fracture at fracture risk assessment tool (FRAX), and they had a lower BMD of the femoral neck than ACPA-negative RA patients [101].
Autoantibodies in RA have been reported to be associated with osteoporosis not only in experimental studies, but also in clinical studies. In particular, association with decreased BMD was reported in the preclinical period of RA without inflammation, suggesting that autoantibodies directly affect bone metabolism (Figure 2).
4) Increased imbalance between bone resorption and bone formation in RA
The RANKL-RANK-OPG pathway plays an important role in bone loss by promoting the differentiation of osteoclast precursors to osteoclasts, whereas the Wnt signaling pathway plays an important role in bone formation through osteoblast maturation. In normal bone remodeling, these pathways are properly balanced through precise coordination with cells and cytokines. However, when this balance is disrupted by various causes, osteoporosis can occur [102,103].
In RA patients, compared with the control group, the serum RANKL level was higher in the patient group, and the OPG level and OPG/RANKL ratio were significantly higher in the control group. This suggests that osteoclastogenesis is further activated through the RANKL-RANK-OPG pathway in RA patients [10]. DKK1 and sclerostin, which are inhibitors of the Wnt pathway, are stimulated in RA by inflammatory cytokines such as TNF-α, which ultimately inhibits bone formation [74,104]. Elevation of DKK1 was observed in the synovial membrane and serum of RA patients and was found to increase the risk of bone erosion [105].
Increased pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, in the inflammatory state of RA contribute to the imbalance of bone formation and bone resorption through activation of the RANKL/RANK/OPG pathway and inhibition of the Wnt pathway. Immune cells, such as macrophages, synovial fibroblasts, T cells, and B cells, which play an important role in the inflammatory process, as well as autoantibodies such as RF and ACPA can also contribute to bone loss through this imbalance [106].
Biologics and osteoporosis in RA
Most conventional synthetic DMARDs (csDMARDs) used in RA can be expected to show a positive effect on bone density and metabolism by reducing inflammation. Nonetheless, evidence of their effectiveness in preventing bone loss is lacking, and results in this area remain controversial. Methotrexate (MTX) showed a non-deleterious effect on BMD, and leflunomide was observed to significantly increase lumbar spine BMD [107,108].
On the other hand, it has been reported that most bDMARDs effectively inhibit osteoclast-mediated bone loss and exert a positive effect on osteoblasts, contributing to stabilization of bone metabolism [15,25,109].
1) TNF-α inhibitors
As mentioned above, TNF-α was found to play an important role in local and systemic bone loss by promoting bone resorption in RA patients.
TNF-α inhibitors are the primary biologics used for RA patients and include monoclonal antibody agents such as infliximab, adalimumab, golimumab, and certolizumab and soluble receptor agents such as etanercept. It has been reported that TNF-α inhibitors have superior effects to csDMARDs in controlling disease activity and inflammation and prevention of joint damage. In animal models, TNF-α inhibition has been shown to improve the negative balance of bone turnover, decrease osteoclasts, and increase osteoblasts [110,111].
Serum osteocalcin, a marker of bone formation, was shown to be lower in premenopausal female RA patients than in the control group, while N-telopeptide and deoxypyridinoline, markers of bone resorption, were high. Changes in these bone markers were correlated with disease activity [112]. It was reported that TNF-α inhibitors increased bone formation markers and reduced bone resorption markers in several clinical studies (Table 3) [113-116]. In addition, TNF-α inhibitors promoted osteoblastogenesis by significantly reducing DKK1, and they inhibited osteoclastogenesis by increasing OPG, decreasing RANKL, and decreasing the RANKL/OPG ratio (Table 3) [105,117-121].
In additional exploratory analyses of the two-year premier study in RA patients, hand bone loss was evaluated with digital X-ray radiogrammetry. The results showed that adalimumab could reduce metacarpal cortical bone loss [122]. In a cohort study of 184 RA patients receiving adalimumab, after one year of treatment, the BMD of the hip and lumbar spine remained stable, while the BMD of the hand decreased by 1.41% (p<0.0001) [123]. This suggests that TNF-α inhibitors are effective in blocking systemic bone loss; however, they may have limitations in inhibiting local bone loss. In a case-control study comparing RA patients treated with infliximab and RA patients treated with MTX, a significant decrease in spinal and femoral neck BMD was observed in the control group; however, both spine and femoral neck BMD were preserved in the infliximab-treated group. The protective effect of infliximab on bone was observed regardless of disease activity [124]. Other studies using TNF-α inhibitors showed similar results [125,126].
2) Abatacept
Abatacept (a cytotoxic T-lymphocyte-associated protein 4 [CTLA4]–immunoglobulin fusion protein) inhibits T cell activation and differentiation by blocking signal transmission between T cells and antigen presentation cells by binding with CD80 or CD86. It plays an important role in pathogenesis-based treatment of RA [127,128].
In an experimental study on murine peripheral blood mononuclear cells, binding of CTLA4 to osteoclast precursor cells directly inhibited osteoclast differentiation and maturation [129]. In CD80/86-deficient mice, increased osteoclast differentiation induced osteopenia, suggesting that CD80/86-targeting abatacept may inhibit osteoclastogenesis [130]. Moreover, it was reported that abatacept directly suppresses osteoclastogenesis by interfering with intracellular calcium oscillations in bone marrow macrophages [131], and it promotes osteoblastogenesis through increased production of Wnt protein in T cells [132,133]. Meanwhile, in a mouse model, abatacept inhibited PTH-induced bone loss [134]. In a clinical study, abatacept significantly reduced serum RANKL [135]. In addition, abatacept significantly elevated serum OPG and significantly decreased serum DKK1 [136]. These results suggest that abatacept may be involved in both inhibition of osteoclastogenesis and promotion of osteoblastogenesis (Table 3).
In the AIRTIGHT study, a prospective, comparative, and non-randomized study, the effects of abatacept and other bDMARDs on bone metabolism were investigated. Abatacept treatment was significantly associated with an increase in femoral neck BMD. The efficacy of abatacept on the increase of femoral neck BMD was superior to that of other bDMARDs. Although the mechanism by which abatacept increases BMD in the femoral neck remains unclear, CTLA4 immunoglobulin is known to promote Wnt-10b production and bone formation [137]. In an interim analysis of a three-year longitudinal cohort study, the protective effects of various biologics and csDMARDs on systemic bone loss were investigated. Compared with csDMARDs and TNF-α inhibitors, abatacept showed a better BMD preservation effect in RA [138]. These differences suggest that T cells may play a key role in the pathogenesis of RA-associated osteoporosis. Autoantibodies are also crucial in the pathogenesis of osteoporosis. Abatacept can inhibit the production of autoantibodies by inhibiting the differentiation of T cell-dependent B cells into plasma cells. On the other hand, bDMARDs targeting pro-inflammatory cytokines were found to have a smaller effect on ACPA level [139,140]. The ability of abatacept to inhibit autoantibody production may also have induced differences between these drugs.
3) Rituximab
Rituximab is a monoclonal antibody against the molecule CD20 on the surface of B cells. Since the CD20 molecule is involved in complement activation, rituximab induces B cell apoptosis through complement-mediated cytotoxicity after binding to CD20. Currently, it is being used in clinical settings with approval as a secondary biologic treatment for RA patients [141,142]. In an RA mouse model, B cells inhibit bone formation through suppression of osteoblast differentiation and functions [143].
In a study analyzing the cytokine mRNA profile of inflammatory cells isolated from the synovial fluid of 12 RA patients, high expression of mRNA for RANKL was observed in B cells. This finding suggests that B cells are the main source of RANKL [144]. In addition, anti-CD20-mediated B cell depletion was related to increased bone mass in a mouse model [145]. In a study using the synovial membranes of 28 RA patients treated with rituximab, RANK-positive osteoclast precursors and synovial RANKL expression were significantly reduced. In serum, OPG and RANKL were significantly decreased, and the OPG/RANKL ratio was significantly increased. These results partially explain the protective effect of rituximab on the progression of bone loss in RA [146].
In an exploratory and prospective study examining changes in bone density and bone turnover in RA patients treated with rituximab, lumbar spine and forearm BMD were effectively maintained, whereas femur BMD was significantly decreased. Furthermore, significant elevations of procollagen type 1 amino-terminal propeptide (P1NP), and bone-specific ALP, all biomarkers of bone formation, were observed (Table 3) [147].
4) IL-6 inhibitor
Blockade of IL-6 receptors directly inhibited osteoclast formation in vitro and in vivo, and this effect was observed regardless of the anti-inflammatory effect of anti-IL-6R therapy [148]. Tocilizumab is a monoclonal antibody that binds to IL-6 receptor, a pro-inflammatory cytokine, and inhibits inflammation-related IL-6 signaling [149]. In a one-year prospective open study, RA patients treated with tocilizumab showed a decrease in serum DKK1 concentration and an increase in serum P1NP, an osteogenic marker, without significant change in BMD [150]. In the radiate study, which was a randomized, double-blind, placebo-controlled, parallel-group phase 3 trial, the effects of tocilizumab on bone metabolism markers in 299 anti-TNF refractory RA patients were investigated. In that study, the tocilizumab group showed significantly reduced C-terminal telopeptides of type 1 collagen (CTX), CTX/osteocalcin ratio, and matrix metalloproteinase 3 compared to the MTX group [151]. A study comparing changes in bone marrow tissue obtained from 10 RA patients treated with tocilizumab and 10 other RA patients treated with MTX monotherapy demonstrated a significant increase in OPG expression in tocilizumab-treated patients compared with the control [152]. In a preliminary study comparing the effects of tocilizumab on bone homeostasis in 22 patients with active RA and 22 healthy patients, the serum level of DKK1 was decreased in the tocilizumab group compared with baseline after two months of treatment. The ratio of OPG/RANKL was significantly increased. These results suggest that the positive effect of tocilizumab on bone remodeling was induced by reduction of the effect of DKK1 on the Wnt pathway and rapid and effective inhibition of inflammation [153].
In a multicenter single-arm study, tocilizumab maintained stable BMD for two years [154]. Another study showed a significant increase of lumbar and femoral neck BMD in patients with osteopenia at baseline [155]. In ACPA-positive RA patients, two years of tocilizumab treatment significantly decreased CTX level and significantly increased femoral neck BMD [156]. In a retrospective study investigating bone metabolism and BMD when denosumab was used in combination with anti-TNF inhibitors, tocilizumab, or abatacept in osteoporotic patients with RA, tocilizumab was more beneficial than the other drugs in increasing BMD in the hip joint [157].
In summary, biologics used for RA induce positive changes in the bone turnover marker profile and show good results in improving BMD (Table 3). Theoretically, it is estimated that abatacept, which is more effective in reducing autoantibody production along with T cell inhibition, would be more advantageous in inhibiting bone loss. However, there does not appear to be a clear difference between the drugs. Abatacept and tocilizumab have been reported to improve BMD better than other biologics in some studies [137,157]; further research is needed in this regard.
CONCLUSION
In this review, we investigated the pathogenesis of osteoporosis associated with RA and the effects of biologics on osteoporosis. In addition to the traditional risk factors for osteoporosis, RA increases the risk of osteoporosis compared to the general population owing to several additional factors, such as chronic inflammation, autoantibodies, drugs, and decreased physical activity. In particular, in RA, immune cells, pro-inflammatory cytokines, and autoantibodies related to autoimmune reactions interact with RANKL-RANK-OPG and Wnt pathways to promote bone resorption and inhibit bone formation. Through these mechanisms, bone loss can be further aggravated in RA. Under this consideration, control of inflammation using biologics in RA may not only inhibit disease progression and improve the quality of life, but also may be important in preventing and treating osteoporosis. In agreement, several studies have reported positive results of biologics in improving BMD with changes in the bone turnover marker profile. However, the effects of these treatments on bone remain largely unknown. Further studies should be conducted on the pathophysiology of osteoporosis associated with RA and the clinical effects of various treatments including biologics.
 XML Download
XML Download