

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Overactive bladder (OAB) is a syndrome characterized by urinary urgency, regardless of urgency urinary incontinence (UUI), and is usually associated with frequency and nocturia [1]. Muscarinic receptor antagonists, such as oxybutynin, solifenacin, darifenacin, fesoterodine, propiverine, and tolterodine, are widely used to treat OAB. Fesoterodine, which was approved by the US Food and Drug Administration in 2008, is a novel antimuscarinic agent that is effective and well-tolerated in symptomatic OAB treatment, including UUI [2,3]. Its administration is more flexible than other muscarinic antagonists [4]. Fesoterodine has limited potential to cross the blood-brain barrier, which is manifested clinically by a low incidence of central nervous system (CNS) adverse effects. Consequently, it is not contraindicated in patients with CNS diseases [5]. Although it is efficacious for the treatment of OAB, such as other muscarinic receptor antagonists, it has several side effects, such as constipation, insomnia, headaches, dry mouth, and blurred vision. In addition to these common side effects, an impact on ion channels has also been reported. For example, fesoterodine inhibits the human ether-a-go-go-related gene (hERG) channel, expressed in human embryonic kidney (HEK293) cells with an IC50 value of 3.6 μM [6]. However, its effects on vascular K+ channels, specifically voltage-dependent K+ (Kv) channels, have not been reported. Therefore, we investigated the effects of fesoterodine on Kv channels using native coronary arterial smooth muscle cells.
K+ channels regulate the resting membrane potential and arterial tone. Their activation causes membrane hyperpolarization and vasorelaxation, while their inhibition causes membrane depolarization and vasoconstriction [7]. Various types of K+ channels, such as ATP-sensitive K+ (KATP), Ca2+-activated K+ (BKCa), inward rectifier K+ (Kir), and Kv channels, have been identified in arterial smooth muscles [7]. Kv channels, which are the most common type in vascular smooth muscles, maintain the resting membrane potential, and as a result, the vascular resting tone [8,9]. Furthermore, functional changes in Kv channels have been implicated in the pathogenesis of various cardiovascular and metabolic diseases [10-12]. In fact, the use of Kv channel inhibitors leads to membrane depolarization and vasoconstriction [13,14]. In addition, Kv channel activity in arterial smooth muscles is reduced in patients with various cardiovascular and metabolic diseases [9,15]. Therefore, the effects of drugs on Kv channels should be clearly elucidated to avoid problems in arterial function.
In the current study, we investigated the inhibitory effect of fesoterodine, a muscarinic receptor antagonist, on arterial Kv channels. Our results suggest that fesoterodine inhibits arterial Kv channels in a concentration- and use (state)-dependent manner, independent of its anti-muscarinic function.
METHODS
Single cell isolation
All animal experimental procedures were performed in accordance with the guidelines of the Committee for Animal Experiments at Kangwon National University (approval No. KW-210512–1). Male New Zealand white rabbits (age: 8–9 weeks; weight: 2.0–2.2 kg) were obtained from Nara Biotech Co., Ltd. (Seoul, Korea). The rabbits were euthanized with an overdose of pentobarbitone (40 mg/kg) and heparin (100 U/kg) administered into the ear vein. The hearts were rapidly removed and placed in ice-cold Normal Tyrode’s (NT) solution, and the coronary artery was carefully isolated from the heart using a microscope. The arteries were mechanically deendothelialized by introducing air bubbles into the arterial lumen. Single smooth muscle cells were obtained through a two-step enzymatic procedure. First, the coronary arteries were digested in Ca2+-free NT solution containing 1.3 mg/ml bovine serum albumin, 1.8 mg/ml dithiothreitol, and 1.2 mg/ml papain for 22–23 min at 37.5°C. Next, the tissues were transferred to a Ca2+-free NT solution containing 2.5 mg/ml collagenase with the same concentrations of bovine serum albumin and dithiothreitol for 16–17 min at 37.5°C. Subsequently, the digested arteries were agitated softly to disperse the cells using Kraft–Brühe (KB) solution. The cell suspension was stored at 4°C in KB solution and used within 4 h.
Solutions and drugs
The standard NT solution contained the following (in mM): NaCl 137, KCl 4.4, CaCl2 1.7, MgCl2 0.6, glucose 13, HEPES 7, and NaH2PO4 1.1. pH was corrected to 7.4 with NaOH. The whole-cell patch pipette solution contained the following (in mM): K-aspartate 118, KCl 28, EGTA 15, HEPES 10, Mg-ATP 3.8, NaCl 5.2, and MgCl2 1.5, buffered to pH 7.25 with KOH. The KB solution contained the following (in mM): KCl 48, KOH 73, KH2PO4 23, L-glutamate 52, HEPES 10, glucose 17, MgCl2 3.5, EGTA 3.5, and taurine 17, titrated to pH 7.3 with KOH. Fesoterodine was purchased from APExBIO (Apexbio Technology LLC, Houston, TX, USA) and prepared in dimethyl sulfoxide (DMSO). DPO-1, guangxitoxin, and linopirdine were purchased from Tocris Cookson (Ellisville, MO, USA). linopirdine was dissolved in water, and the others were dissolved in DMSO. The final concentration of DMSO was less than 0.1% volume, and this concentration did not affect either the membrane currents or resting membrane potential.
Electrophysiological recordings and data analysis
In whole-cell patch clamp experiments, the Kv currents of single smooth muscle cell were measured using a patch-clamp amplifier (EPC-8; Medical System Corp., Darmstadt, Germany) and digital interface adapter (NI-DAQ-7; National Instruments, Union, CA, USA). Data acquisition and generation of various voltage pulses were performed using the PatchPro software. Patch pipettes with resistances of 3–4 MΩ were pulled from borosilicate glass capillaries (Clark Electromedical Instruments, Reading, UK) using a microelectrode vertical puller (PP-830; Narishige, Tokyo, Japan). The approach of series resistance was less than 10 MΩ in the whole-cell configuration. The voltage error from the voltage drop at the series resistance was calculated to be ~4 mV. No rectification was made for this voltage error. The average cell capacitance was 15.71 ± 0.33 pF (n = 18). All electrophysiological recordings were performed at room temperature (20°C–25°C).
Analysis of data was performed using Origin ver. 8.0 software (Microcal Software, Inc., Northampton, MA, USA). A concentration-response curve for Kv current inhibition by fesoterodine was fitted to the following Hill equation:
where ƒ is the percentage block of the Kv current (ƒ(%) = [1 – Idrug ÷ Icontrol] × 100) at the test voltage, [D] is the fesoterodine concentration, IC50 is a half-maximal inhibitory concentration, and n is the Hill coefficient.
Activation curves were derived from tail currents produced by reverting to a potential of –40 mV based on short depolarizing pulses (20–50 ms) from –80 to +60 mV in the absence and presence of fesoterodine. The recorded tail currents were normalized to the maximal tail currents. Activation curves were fitted using the following Boltzmann equation:
where k, V, and V1/2 represent slope value, preconditioning test potential, and medium-point of activation potential, respectively.
A steady-state inactivation curve was obtained from the following voltage protocol. The Kv currents were recorded by applying a test potential (+60 mV) after preconditioning steps of –80 to +30 mV for 7 sec in the absence and presence of fesoterodine. Inactivation curves were elicited using another Boltzmann equation:
where k, V, and V1/2 represent slope value, preconditioning test potential, and medium-point of inactivation potential, respectively.
RESULTS
Fesoterodine inhibits Kv channels in coronary arterial smooth muscle cells
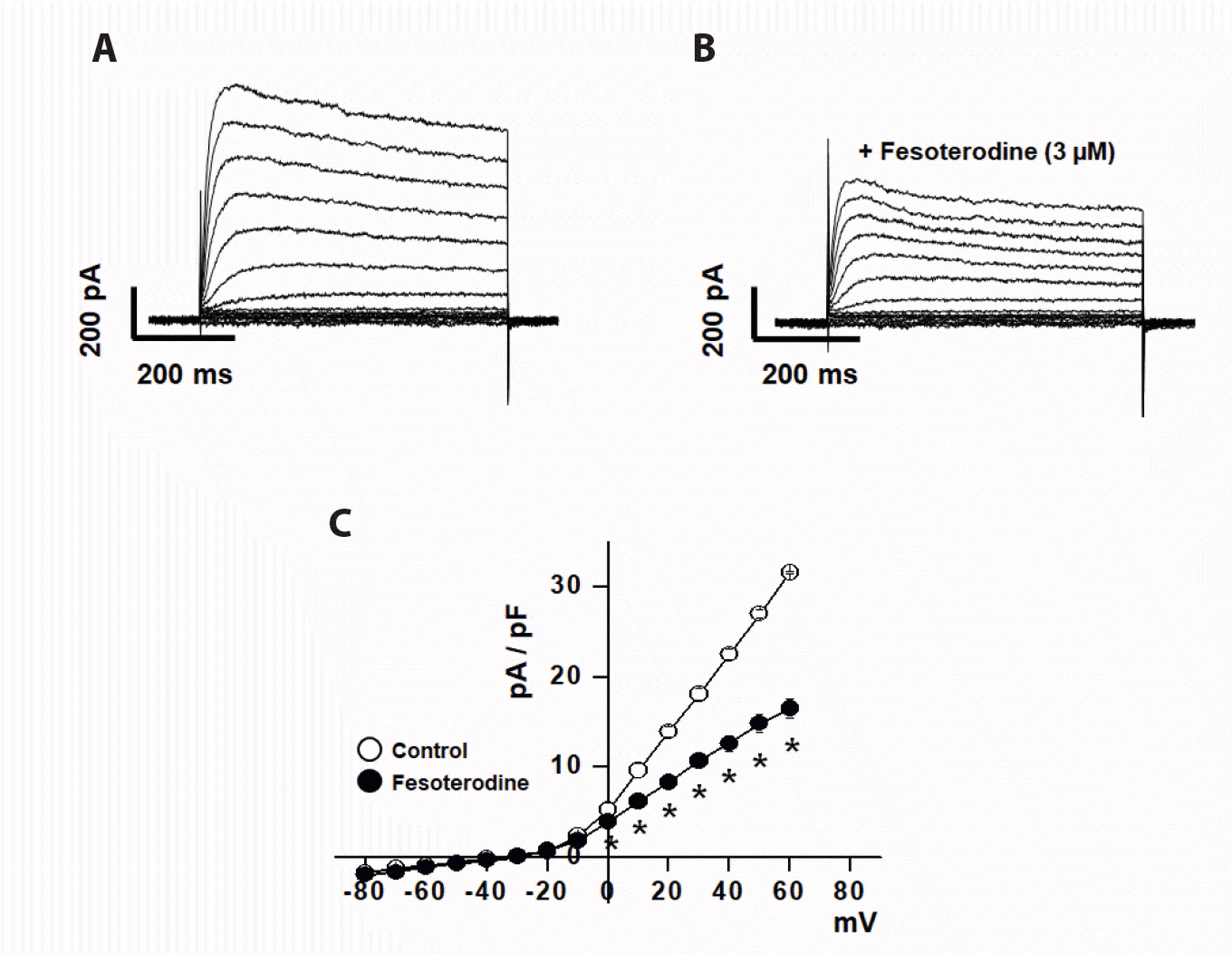
Single smooth muscle cells were voltage-clamped to a holding potential of –80 mV and 600 ms depolarizing pulses from –80 to +60 mV were applied every 15 sec to activate the Kv currents (Fig. 1A). The currents were rapidly activated to a peak, followed by slow and partial inactivation. The activation of other vascular K+ channels was excluded by including 3.8 mM ATP to block KATP channels and 10 mM EGTA to block BKCa channels in the pipette solution. The involvement of Kir channels was also excluded using large-diameter (conduit) arteries, which do not express Kir channels. Superfusion with fesoterodine (3 μM) inhibited the Kv currents (Fig. 1B). Fesoterodine-induced Kv current inhibition occurred immediately and reached a steady-state within 1 min. Fig. 1C shows the steady-state current-voltage (I–V) relationships in the absence and presence of 3 μM fesoterodine. The I–V relationship shifted significantly downward over the full voltage range at which the Kv channels were activated. For example, 3 μM fesoterodine inhibited Kv currents by 49% at 60 mV.
Concentration-dependent vascular Kv channel inhibition by fesoterodine
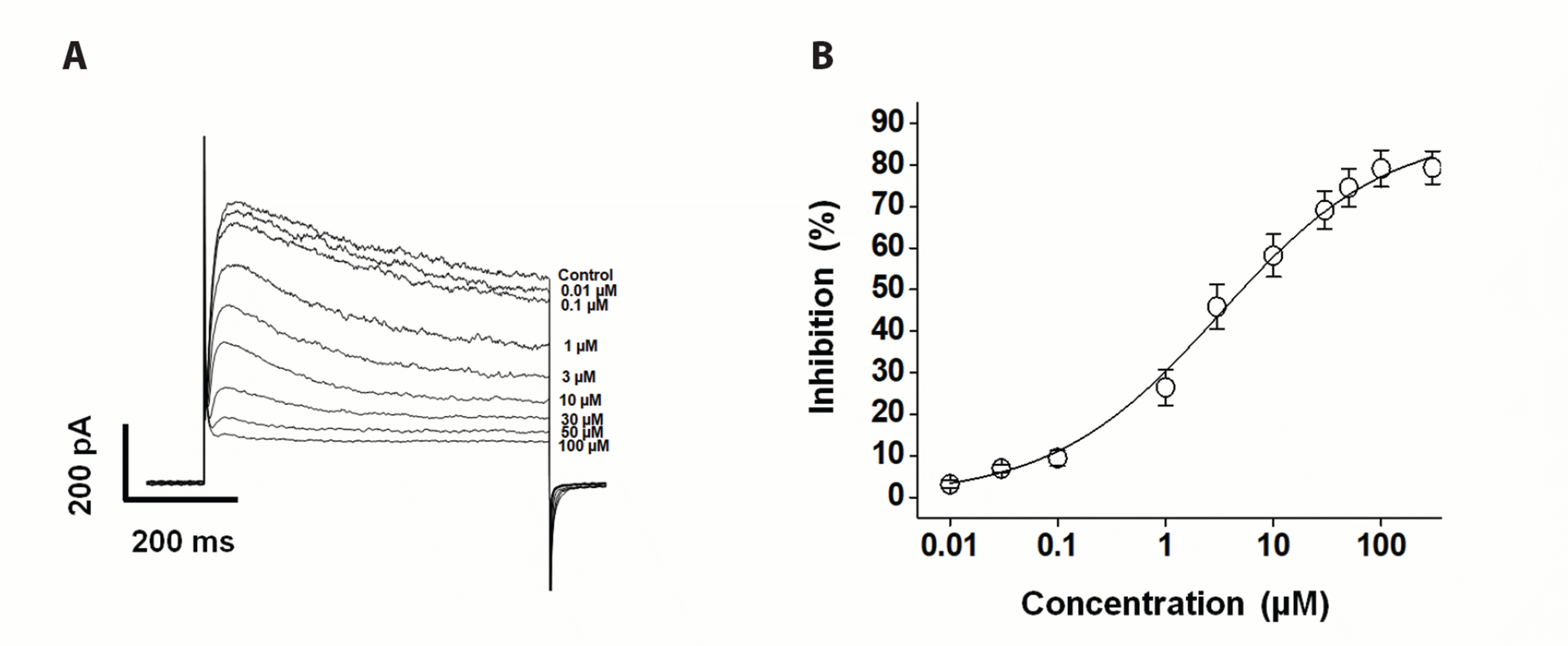
Fig. 2 shows the superimposed Kv currents under control conditions and in the presence of various fesoterodine concentrations (0.01, 0.1, 1, 3, 10, 30, 50, and 100 μM). The Kv currents were induced by applying a one-step depolarization pulse of +60 mV at a holding potential of –80 mV. The inhibitory effect of fesoterodine on the Kv currents was augmented by increasing its concentration (Fig. 2A). The concentration-dependent Kv current inhibition was measured at the end of a depolarizing pulse of +60 mV, and was normalized to the Kv current recorded in the absence of fesoterodine. Data were fitted to the Hill equation, yielding an IC50 value of 3.19 ± 0.91 μM and a Hill coefficient of 0.56 ± 0.03 (Fig. 2B). These results indicate that fesoterodine induces Kv channel inhibition in a concentration-dependent manner.
Effect of fesoterodine on steady-state activation and inactivation properties of Kv channels
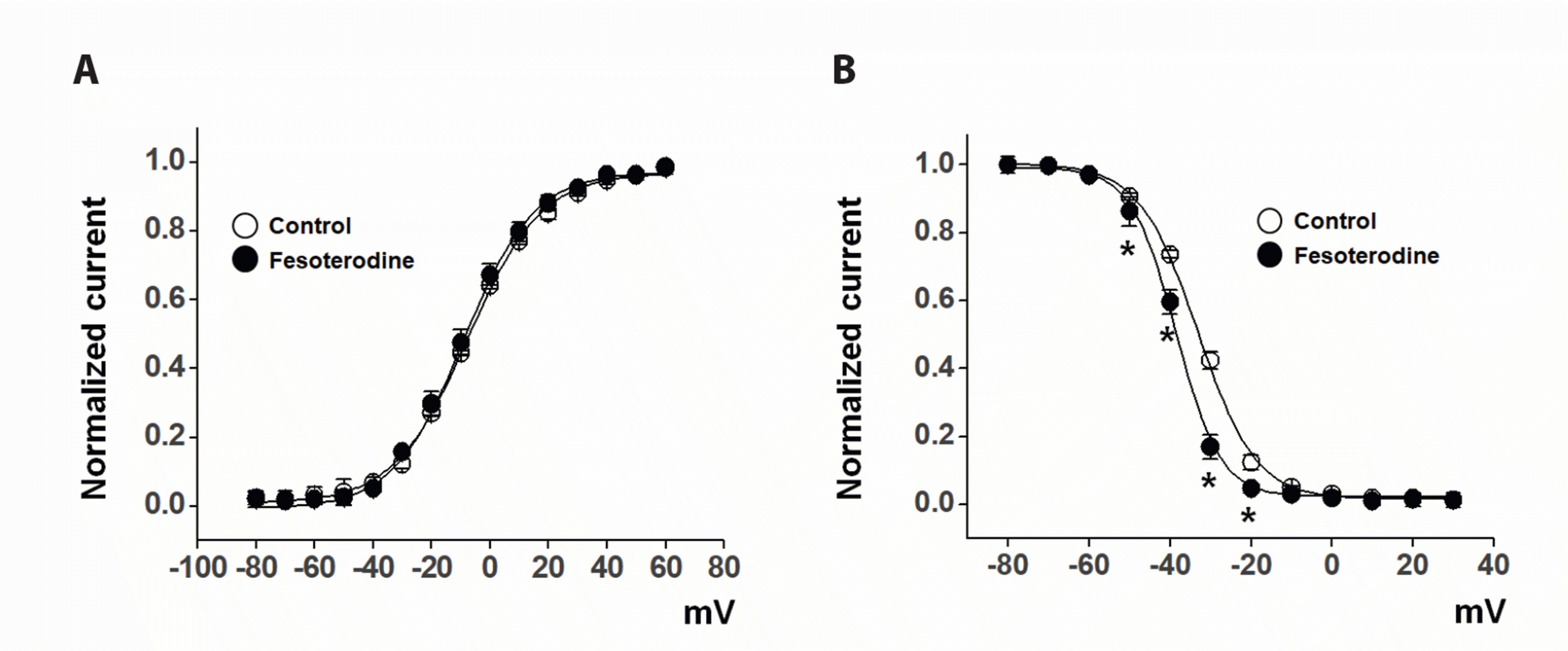
To determine whether fesoterodine-induced Kv current inhibition occurred by affecting channel gating properties, changes in the steady-state activation and inactivation curves were observed before and after fesoterodine administration. The activation curves were acquired by analyzing the tail currents of a typical protocol, as described in Materials and Methods, and fitted to the Boltzmann equation. Fesoterodine (3 μM) did not change the curve (Fig. 3A). The medium-point of the activation potential (V1/2) and slope value (k) were –7.67 ± 0.39 mV and 12.62 ± 0.41 under the control conditions, and –8.59 ± 0.64 mV and 12.06 ± 0.60 in the presence of 3 μM fesoterodine, respectively.
The inactivation curves were also obtained using the typical double-pulse protocol, described in Materials and Methods, and fitted to another Boltzmann equation. Fesoterodine (3 μM) induced a significant negative shift in the curve (Fig. 3B). The medium-point of the inactivation potential (V1/2) and slope value (k) were –33.36 ± 0.44 mV and 7.02 ± 0.32 under the control conditions, and –38.21 ± 0.59 mV and 5.45 ± 0.45 in the presence of 3 μM fesoterodine, respectively. These results suggest that fesoterodine inhibits Kv channels by affecting their gating properties.
Use (state)-dependent block of Kv currents by fesoterodine
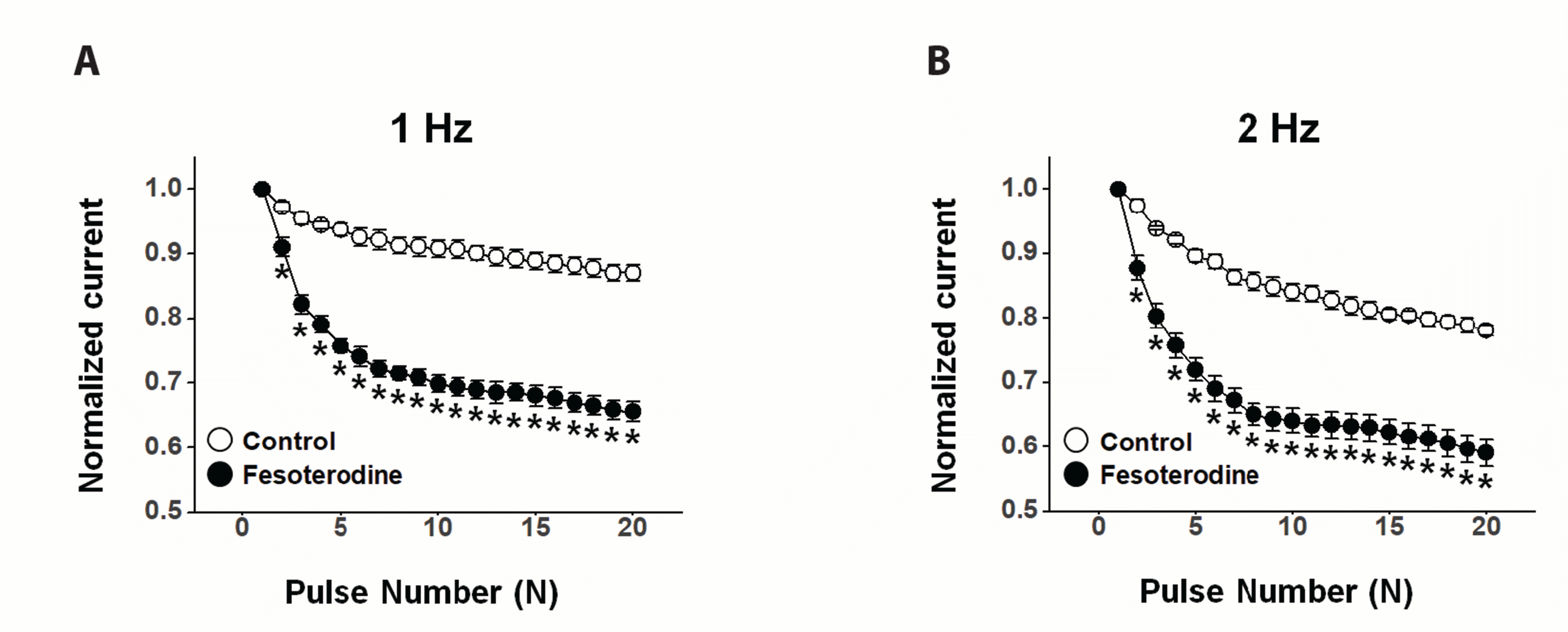
To test whether the fesoterodine-induced Kv channel inhibition was related to the channel state, 20 repetitive depolarizing pulses of +60 mV from a holding potential of –80 mV were applied at frequencies of 1 or 2 Hz in the absence and presence of 3 μM fesoterodine. The peak amplitudes of the Kv current decreased by 13% at a frequency of 1 Hz and by 22% at a frequency of 2 Hz under control conditions (Fig. 4). In the presence of 3 μM fesoterodine, the peak amplitude of the Kv current progressively decreased by 34% and 41% at 1 and 2 Hz, respectively. This strong frequency dependence suggests that Kv current inhibition by fesoterodine occurs in a use (state)-dependent manner.
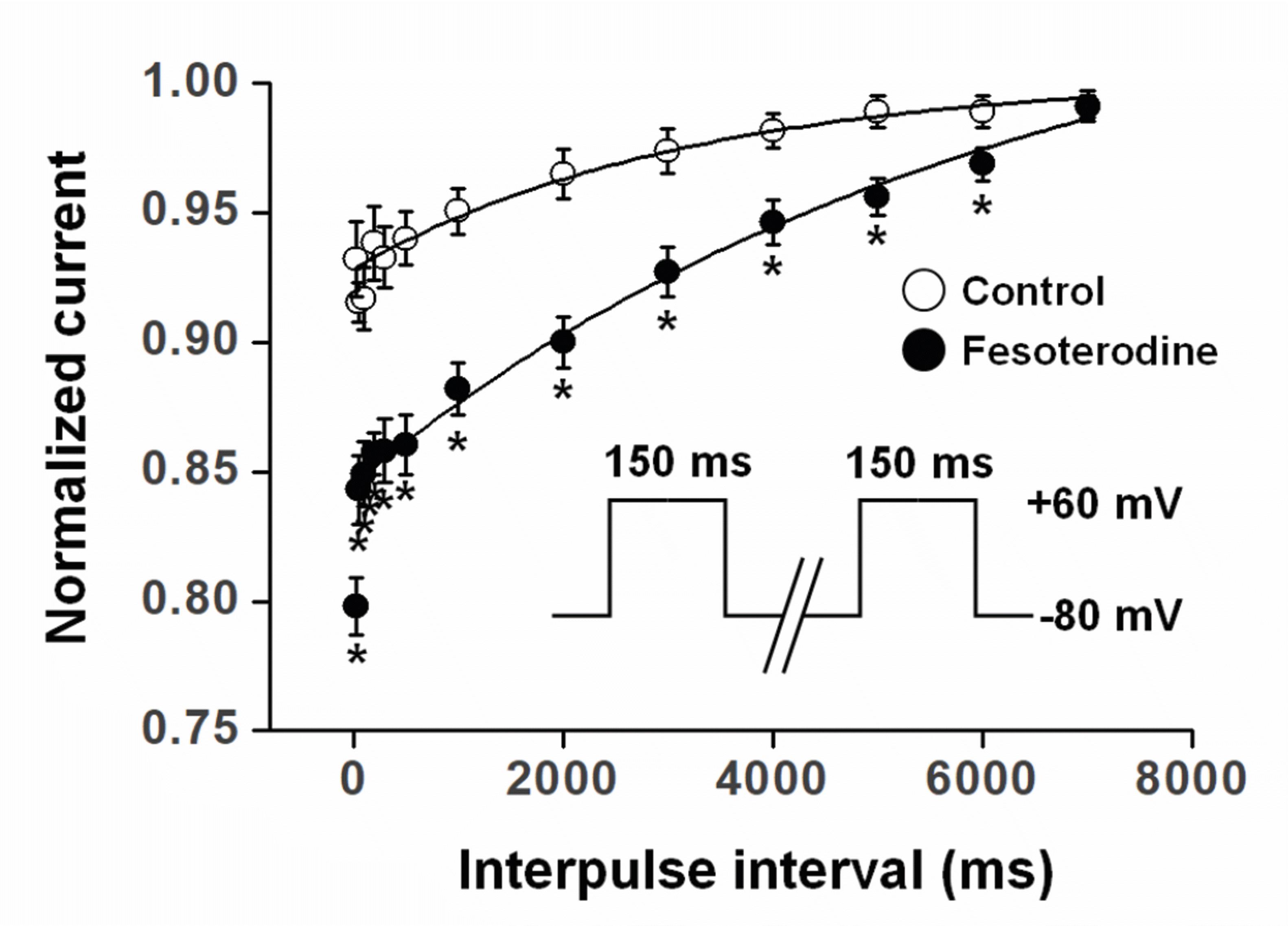
To further investigate the use (state)-dependent Kv current inhibition by fesoterodine, the recovery time from channel inactivation was measured by applying identical pulses with various interpulse intervals in the absence and presence of 3 μM fesoterodine. The interval between the two identical pulses increased gradually from 30 ms to 7,000 ms. Fesoterodine significantly increased the recovery time constant (Fig. 5), which was 3,201.1 ± 218.8 ms under the control conditions and 6,136.7 ± 399.4 ms in presence of fesoterodine. The slowed constant in the presence of fesoterodine suggests that fesoterodine inhibits Kv channels in a use (state)-dependent manner.
Effect of fesoterodine on Kv1.5, Kv2.1 and Kv7 subtypes
Several Kv channel subtypes have been identified in vascular smooth muscle cells [16]. Among these, Kv1.5, Kv2.1, and Kv7 are the major subtypes in vascular smooth muscles, and their inhibitors are commercially available. Therefore, we investigated the role of these subtypes in the inhibitory effect of fesoterodine. Pretreatment with the Kv1.5 subtype inhibitor DPO-1 (1 μM) reduced the Kv currents, but subsequent application of 3 μM fesoterodine did not induce further inhibition of Kv currents (Fig. 6A, B), suggesting that fesoterodine-induced inhibition of Kv currents is strongly associated with Kv1.5 inhibition. Pretreatment with the Kv2.1 inhibitor guangxitoxin (30 nM) and the Kv7 inhibitor linopirdine (10 μM) also inhibited the Kv currents. However, subsequent application of 3 μM fesoterodine inhibited the Kv currents at levels similar to fesoterodine application alone (Fig. 6C–F). These results suggest that the Kv2.1 and Kv7 subtypes were not involved in the inhibitory effect.
DISCUSSION
The present study demonstrated that fesoterodine causes concentration- and use (state)-dependent inhibition of Kv channel currents regardless of muscarinic receptor antagonism. Furthermore, it shifts the steady-state inactivation curve to a more negative potential without affecting the activation curve.
Fesoterodine-induced Kv current inhibition occurred in a state-dependent manner, evidenced as follows. First, Kv channel inhibition gradually increased during the application of train pulses in the presence of fesoterodine (Fig. 4). Furthermore, fesoterodine prolonged the recovery time constant from Kv channel inactivation (Fig. 5). These results suggest that the blocking action of fesoterodine is use (state)-dependent. Second, fesoterodine shifted the voltage-dependent inactivation curve (Fig. 3). Although changes in activation and/or inactivation by some drugs are not confirming evidence of state-dependent inhibition, most drugs that do this affect steady-state activation and inactivation kinetics [17,18]. In general, state-dependent channel inhibition is classified into open and closed states. A characteristic of open-state inhibition is that the drug predominantly inhibits the steady-state portion, more than the peak of Kv currents. However, fesoterodine suppressed the peak and steady-state amplitudes of the Kv currents to a similar extent, suggesting that Kv current inhibition by fesoterodine occurred in a closed-state of the channels (Figs. 1 and 2). This needs further investigation through structural binding analysis between channels and drugs.
The inhibitory effect of fesoterodine on Kv channels occurs independently of muscarinic receptor inhibition, as the inhibition of Kv currents began immediately after fesoterodine administration and reached a steady-state within 1 min. In fact, fesoterodine as a nonselective muscarinic receptor antagonist regulates smooth muscle tone through cellular electrical signals by preventing the binding of acetylcholine to muscarinic acetylcholine receptors [3]. However, the rapid inhibitory response of fesoterodine on Kv channels suggests a direct interaction between fesoterodine and Kv channels rather than through a complex signaling pathway. Furthermore, fesoterodine inhibited Kv channels in a state-dependent manner (Figs. 4 and 5), which suggests that it directly interacts with the channels or binds near them. Either case may occur regardless of muscarinic receptor inhibition. The steady-state inactivation curve provides further evidence of the lack of involvement of muscarinic receptor inhibition in the effects of fesoterodine. In fact, the effect on the activation and/or inactivation curve by some drugs suggests that these drugs could interact with voltage sensor of channels or bind near the channels. In addition, the IC50 value of fesoterodine for Kv channel inhibition was 3.19 ± 0.91 μM, different from the value for M3 receptor antagonists (7.4 nM) [19]. The distinct discrepancy in these values also indicates that fesoterodine inhibits Kv channels independently of muscarinic receptor inhibition. Furthermore, previous studies in our group demonstrated that an alternative anti-muscarinic drug atropine had no significant effect on the Kv current. These results are also strong evidence that fesoterodine effect was not associated with anti-muscarinic action [20-22]. Based on this evidence, fesoterodine clearly interacts with Kv channels regardless of muscarinic receptor antagonism.
Kv channels expressed in vascular smooth muscle cells are major regulators of vascular tone and resting membrane potential. Therefore, it is important to identify the effects of drugs on the vascular Kv channels in vascular functional studies [16,23]. Indeed, several antimuscarinic drugs inhibit vascular Kv channels. Another muscarinic receptor antagonist, tolterodine, inhibits vascular Kv channels in a concentration-, time-, and use (state)-dependent manner with an IC50 value of 1.71 μM [20]. Like fesoterodine, tolterodine shifts the steady-state inactivation curve towards a negative potential. The anti-muscarinic drug oxybutynin also inhibits vascular Kv channels by shifting both activation and inactivation curves with an IC50 value of 11.51 μM [21]. More recently, darifenacin, an anticholinergic agent, was found to inhibit vascular Kv channels in a concentration- and use (state)-dependent manner by shifting both the activation and inactivation curves [22]. Some drugs used for OAB syndrome treatment also inhibit vascular Kv channels. Based on these findings, drugs for OAB treatment, including fesoterodine, should be prescribed carefully, particularly in patients with vascular diseases.
Many Kv channel subtypes (Kv1–Kv12) have been detected in vascular smooth muscle cells. Although the expression of Kv subtypes varies among species, Kv1, Kv2, and Kv7 are abundantly expressed in vascular smooth muscles [7,9]. However, studies of the expression of Kv subtypes have been performed mainly in mice, rats, and humans. Our study investigated Kv channels using rabbit arteries because their cardiovascular properties and structure are similar to humans [24]. The exact Kv subtypes expressed in rabbit arteries are still uncertain. However, previous studies have reported the expression of Kv1.5, Kv2.1, and Kv7 in rabbit arterial smooth muscles [25-30]. Furthermore, administration of Kv1.5, Kv2.1, and Kv7 inhibitors effectively decreased the Kv current (Fig. 6), indicating that these subtypes are expressed in rabbit arteries. Our results demonstrate that fesoterodine application in the presence of a Kv1.5 inhibitor reduced inhibition of steady-state Kv current, suggesting that the primary targets of fesoterodine on Kv channels are Kv1.5. Due to the lack of some specific Kv subtype inhibitors and information on Kv subtype expression in rabbit arteries, we were limited to the conclusion that Kv1.5 is the major subtype involved in fesoterodine-induced Kv channel inhibition. However, even in the presence of a Kv1.5 inhibitor, additional application of fesoterodine induced further inhibition of the peak Kv current (Fig. 6A), suggesting that other Kv subtypes may be involved in the effect. This issue should be further investigated using specific expression systems.
In conclusion, we investigated the inhibitory effects of fesoterodine on Kv channels using rabbit coronary artery smooth muscle cells. Fesoterodine inhibited Kv channels (mainly the Kv1.5 subtype) in a concentration- and use (state)-dependent manner by altering the inactivation curve, independent of muscarinic receptor antagonism. Given the clinical efficacy of fesoterodine, caution should be exercised while prescribing it in patients with OAB.
 XML Download
XML Download