

 PDF
PDF Citation
Citation Print
Print


Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by inflammation of the skin, lungs, joints, kidney, and heart (1). Cardiopathy is a common complication in patients with SLE. Lupus myocarditis occurs in 5∼10% of all patients with SLE. Heart failure (HF) of SLE can be resulted from various complications involving heart valves, myocardium, and coronary vessels (2). SLE morbidity and mortality are attributed mainly to cardiopathy. Premature death in patients with SLE is caused mainly by cardiovascular events (3). Though knowledge of the mechanisms and treatments for SLE are improving, the causes of cardiovascular mortality associated with it are poorly understood (4). Abnormal chronic immune responses might increase the risks of cardiovascular risk factors and atherosclerosis, and eventually induce HF in patients with SLE (5). Prevention of irreversible tissue damage is now considered a critical therapeutic goal in patients with SLE. Hence, an effective modeling platform will be required for future research on SLE (6).
Many systemic autoimmune diseases are characterized by the formation of autoantibodies against intracellular autoantigens (7). In SLE, increases in the levels of autoantibodies and autoantibody-derived immune complexes depositing in various tissues increase the risk of SLE pathogenesis and might eventually induce HF (8). Anti-Ro autoantibodies are among the most detected autoantibodies against extractable nuclear antigens closely associated with SLE (9, 10). Anti-La and anti-Sm antibodies are also prevalent in patients with SLE (11). Anti-Ro autoantibodies affect the hearts of patients with SLE. Cardiac damage may also be caused by Ro antigens in fetal heart tissue between weeks 18 and 24 (12, 13). A previous study suggested that anti-Ro antibodies bind apoptotic cardiomyocytes (CMs) and prevent their removal by healthy CMs during cell deletion in embryogenesis (14). Adult patients with connective tissue disease who are positive for anti-Ro antibodies present with abnormal cardiac repolarization (7). However, SLE is a heterogeneous autoimmune disease and these biomarkers do not fully explain its activity (15). Patients with SLE may exhibit active symptoms in the presence or absence of autoantibodies, which suggest that other biochemical pathways are involved (16). Nevertheless, anti-Ro autoantibodies are closely associated with cardiac tissue damage in patients with SLE, and research on this mechanism may help demystify this disease.
In recent years, stem cell-based research has been commonly used in disease modeling. Human-induced pluripotent stem cells (iPSCs) are generated from various patient-derived somatic cells including skin fibroblasts and blood cells (17). The development of iPSCs has provided an innovative tool for disease modeling, drug screening, and regenerative medicine research (18). Human iPSCs and embryonic stem cells (ESCs) have similar characteristics such as self-renewal, pluripotency, and unlimited proliferation. However, use of the former bypasses the ethical issues associated with the use of the latter. Therefore, it is anticipated that patient-derived iPSCs will be more broadly used in various fields in the future. As iPSCs can be generated from virtually any cell type in any individual, they can be applied towards research on heterogeneous diseases such as SLE (19). Also, iPSCs may differentiate into various cell types including those too difficult to harvest for research purposes. Therefore, they might be effective tools for autoimmune disease and cardiac research.
In the present study, we generated and applied healthy control (HC) and SLE patient-derived iPSCs to differentiate CMs. Using a two-dimensional (2D) differentiation platform, we compared the characteristics of HC-iPSC-derived CMs and SLE-iPSC-derived CMs. We treated the differentiated CMs with serum obtained from patients with SLE and analyzed the differences among the latter. Three-dimensional (3D) platforms for cardiac differentia-tion were applied using HC-iPSCs. The impact of SLE serum and anti-Ro autoantibodies on HC-iPSC-derived CMs was confirmed by the altered expression of fibrosis and apoptosis markers. The aims of the present study were to develop and test a proof-of-concept disease-in-a-dish platform using iPSCs and determine the effects of SLE serum and anti-Ro antibodies on CMs. It is hoped that the suggested platform might be helpful in studying the conditions of cardiomyocytes in SLE patients.
Materials and Methods
Patient consent and ethical procedures
Written informed consent was obtained from all partici-pants. The study was approved by the Institutional Review Board/Ethics Committee of the Catholic University of Korea, Seoul, Republic of Korea (No. KC19SNSI0819).
Human iPSC generation using sendai viral vector
To generate iPSCs, peripheral blood mononuclear cells (PBMCs) were obtained from HC and patients with SLE. Blood was delivered in heparin tubes. Fresh blood was diluted with phosphate buffered saline (PBS) and placed onto Ficoll-paque reagent. The samples were centrifuged for 30 minutes at 2,000 rpm. Isolated PBMCs were transferred to a new tube and washed with PBS. Human iPSCs were generated using the method described by Rim et al. (17). PBMCs were cultured for 5 d in StemSpan medium (StemCell Technologies, Vancouver, BC, Canada) to a density of 3×105/well. The cells were then added with Sendai viral vector (Thermo Fisher Scientific, Waltham, MA, USA) and centrifuged at 1,160×g for 30 min. The Sendai viral vector contains Yamanaka factors including OCT4, SOX2, cMyc, and Klf4 to induced reprogramming. The transduced cells were transferred to a 24-well coated with vitronectin (Thermo Fisher Scientific), and centrifuged at 1,160×g for 10 min. The media was changed with Essential 8 media (E8 media, Thermo Fisher Scientific) daily, and attached cells were expanded. Colony forming iPSCs were purified by colony picking. The purified final form of iPSCs were maintained in vitronectin-coated dishes with daily change of E8 media.
2D CM differentiation from iPSCs
CMs were differentiated according to a previously reported method (20). HC-iPSCs and SLE-iPSCs were subcultured in vitronectin-coated plates at 37℃ and under a 10% CO2 atmosphere. Human iPSCs at 90% confluency were incubated for 2 d in differentiation cardiomyocyte differentiation media 3 (CDM3) medium supplemented with 6 μM CHIR99021 (Biogems, CA, USA) also containing 213 μg/ml L-ascorbic acid-2-phosphate and 500 μg/ml human albumin (Sigma-Aldrich, Darmstadt, Germany). On day 3, the Wnt C-59 inhibitor (Selleck Chemicals LLC, Houston, TX, USA) in the medium was replaced with differentiation basal medium for 2 d. After 48 h (day 15 of differentiation), the medium was replaced with inhibitor-free basal medium.
3D CM differentiation from iPSCs
The first 4 days of 3D CM differentiation were identical to those of 2D CM differentiation. Beating in differentiated CMs was observed between days 6 and 8 of differentiation. On day 10, beating cells were dissociated with CM dissociation medium (StemCell Technologies, Vancouver, BC, Canada). A 2:1 mixture of CDM3 and Aggrewell media (StemCell Technologies, Vancouver, BC, Canada) was added to each well containing 4×104 CMs. Spheroids were formed and maintained in a U-bottom 96-well plate (TPP, Schaffhausen, Switzerland) for 24 h. Then 10 μM Rho-associated kinase (ROCK) inhibitor (ROCKi) was added. The next day and the medium were replaced with CDM3 medium every 2 days for 10 days.
Real time-polymerase chain reaction (RT-PCR)
Cells or spheroids were harvested, snap-frozen at −80℃ in liquid nitrogen, and ground in a mortar. The samples were incubated with TRIzol reagent (Thermo Fisher Scientific). Their mRNA was extracted according to the manufacturer’s instructions. A RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used for cDNA synthesis. Approximately 2 μg extracted total RNA was used to synthesize cDNA. RT-PCR was performed using a LightCyclerⓇ 480 Instrument II (Roche Diagnostics, Basel, Switzerland). GAPDH was the internal control for normalization of gene expression and the relative quantification results are shown as 2^-(delta CT) values. Primer sequences are listed in Table 1.
Immunofluorescence staining of iPSCs and 2D CMs
For hiPSC colony formation, 2×103 iPSCs were seeded in a single well of a vitronectin-coated six-well plate. The cells were expanded with E8 medium for 5∼7 d and the medium were replaced daily. The cells were washed with phosphate-buffered saline (PBS) and fixed with 4% (v/v) paraformaldehyde (PFA; Biosesang, Seongnam, Republic of Korea). After another PBS wash, the cells were permeabilized with 0.1% Triton X-100 (Biosesang, Seongnam, Republic of Korea) for 10 min. The cells were then blocked at room temperature (RT) for 30 min using PBS supplemented with 2% (v/v) bovine serum albumin (BSA; Sigma-Aldrich Corp., St. Louis, MO, USA). Primary antibodies were diluted with a mixture of PBS plus BSA (PBA) and incubated at room temperature for 2 h. The following dilution ratios were used: 1/200 anti-SSEA4 (EMD Millipore, Billerica, MA, USA), 1/100 anti-TRA- 1-60 (EMD Millipore, Billerica, MA, USA), 1/100 anti-TRA-1-81 (EMD Millipore, Billerica, MA, USA), 1/100 anti-OCT4 (Santa Cruz Biotechnology, Dallas, TX, USA), 1/100 anti-SOX2 (BioLegend, San Diego, CA, USA), and 1/250 anti-KLF4 (Abcam, Cambridge, UK). After washing the cells, 1/400 Alexa Fluor 594 (Life Technologies, Carlsbad, CA, USA) and 1/400 Alexa Fluor 488 (Life Technologies, Carlsbad, CA, USA) conjugated secondary antibodies were diluted in PBA and incubated at RT in the dark for 1 h. The cells were washed and mounted with ProLong Antifade Mounting Reagent (Thermo Fisher Scientific, Waltham, MA, USA). Colonies were detected by immunofluorescence microscopy.
Immunofluorescence staining was performed with anti- troponin T cardiac isoform (TNNT2) antibody (Invitrogen, Carlsbad, CA, USA) to detect cardiogenic cell markers in the 2D CMs.
Histological analysis of 3D CMs and EB
The 3D CMs and EB (Embryonic Body) of negative control were washed with PBS and fixed in 4% (v/v) paraformaldehyde (PFA) at room temperature for 2 h. The samples were dehydrated with an ethanol gradient series (Biosesang, Seongnam, Republic of Korea). Samples were cleared with sequential ethanol-xylene mixtures and the spheroids were infiltrated with paraffin overnight. The paraffin blocks were fixed and 5-μm sections were prepared with a microtome. The slides were heated in an oven at 60℃ for at least 10 min before staining. The 3D CMs sections were incubated with 10% (v/v) normal goat serum containing 1% PBA to remove any nonspecific background staining. Anti-TNNT2 antibody was diluted in 5% (v/v) normal goat serum containing 1% PBA and incubated at 4℃ overnight. The specimens were incubated with 1/200 Alexa Fluor 488 goat anti-mouse IgG antibody (Life Technologies, Carlsbad, CA, USA) in PBS at RT for 40 min. Nuclear staining was performed with 4’,6-diamidino-2-phenylindole (DAPI).
Autoantibody and human SLE serum treatment
Human serum was collected from two patients with SLE who had provided informed consent (Table 2). We selected the serum of patients that showed a relatively high SLEDAI-2K score (above 6 points). Since the activity of SLE disease varies between individual patients, the sera of the two patients were mixed and used for treatment. CMs differentiated in the presence of small molecules were treated with 10% (v/v) activated SLE serum for 2 d. Each group was treated with 50 μg/ml normal IgG (Santa Cruz Biotechnology, Dallas, TX, USA) or anti-Ro-antibody (Santa Cruz Biotechnology, Dallas, TX, USA) for 2 d.
Cell viability and proliferation assays
To confirm 3D CM spheroid viability, two-color fluorescence analysis and a Live/Dead Viability/Cytotoxicity Assay Kit (Molecular Probes LLC, Eugene, OR, USA) were used according to the manufacturer’s instructions. Proliferation assays were performed on serum-treated differentiated CMs using Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Rockville, MD, USA).
Simultaneous measurement of cytosolic and internal stored Ca2+
The 2D CMs and 3D CMs were washed with PBS and stained with CDM3 medium containing Fluo-4 AM (10 μg/ml; Invitrogen, Carlsbad, CA, USA) in an incubator at 37℃ for 30 min. Then the 2D CMs and 3D CMs were washed with PBS and the CDM3 medium was replaced. The calcium was then measured with a Live Cell Imaging System (Biotek Instruments, Winooski, VT, USA).
Statistical analysis
All experiments were repeated 3∼5 times using three clones per cell line. Data are presented as means±SD. Graphs were generated with GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post- hoc test. T-tests were used for datasets with a normal distribution. Otherwise, Mann-Whitney tests were applied to analyze non-parametric quantitative dataset. Two-tailed p-values were calculated. Statistical significance was indicated as follows: *p<0.05; **p<0.01; ***p<0.001 and ****p<0.0001.
Results
Generation of iPSC from PBMCs of healthy controls and patients with SLE
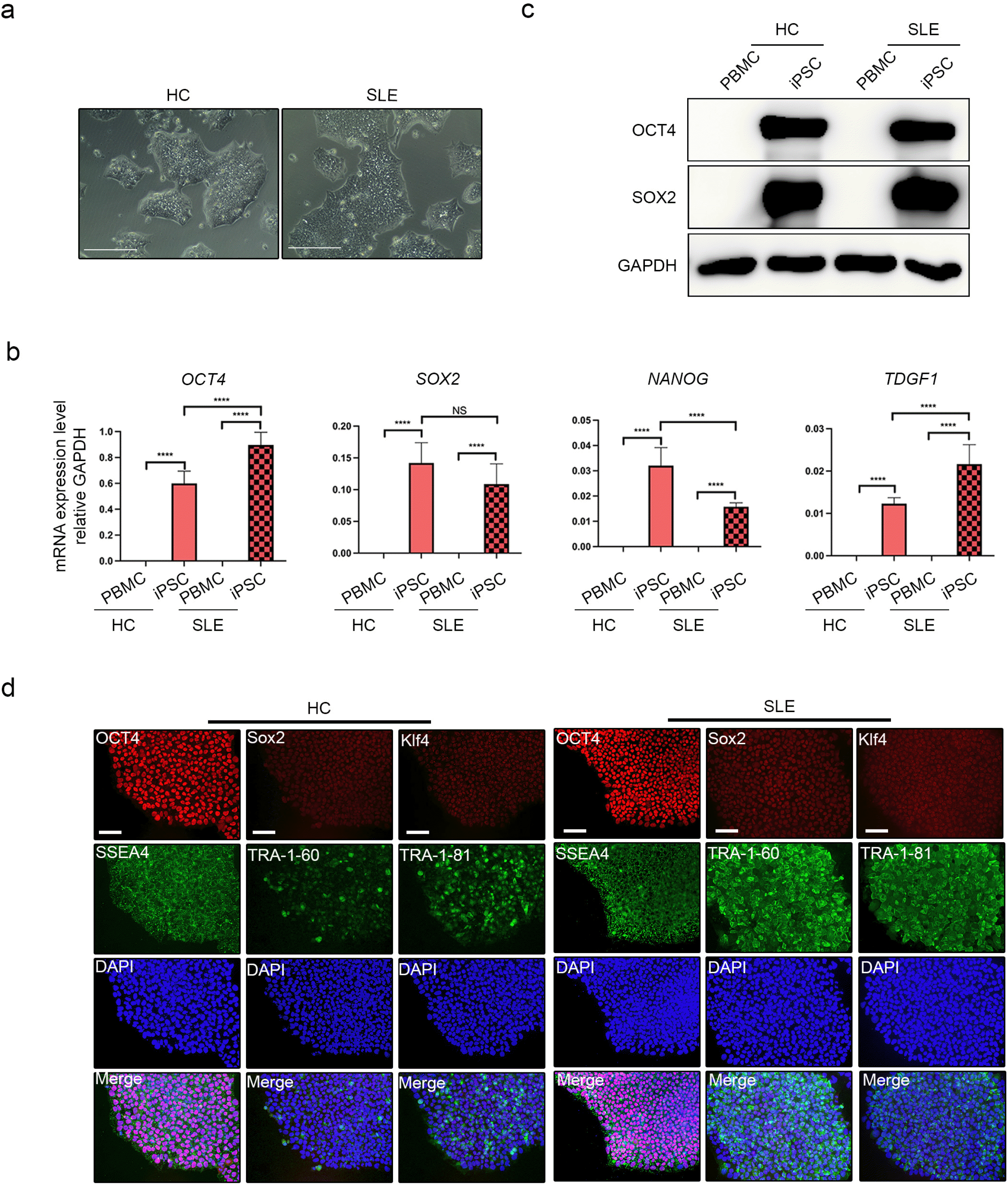
PBMCs from HC and patients with SLE were induced to form iPSCs with Sendai viral vector. Intact colonies were identified in transduced PBMC-derived cells, which is one of the critical characteristics of iPSCs (Fig. 1a). Pluripotent gene (OCT4, SOX2, NANOG, and TDGF1) expression was confirmed in HC-iPSCs and SLE-iPSCs and no significant differences were found between the two group (Fig. 1b). Among the pluripotent markers, the protein expression of OCT4 and SOX2 was confirmed (Fig. 1c). The expression of OCT4 ans SOX2 in the iPSC group of HC and SLE was similar to that of the gene expression. Pluripotent marker expression (OCT4, Sox2, Klf4, SSEA4, TRA-1-60, TRA-1-81) was verified in HC-iPSCs and SLE-iPSCs (Fig. 1d). In conclusion, we confirmed that both iPSC groups exhibited the characteristics of iPSCs, which indicates successful reprogramming.
CM differentiation using iPSCs on 2D culture platform
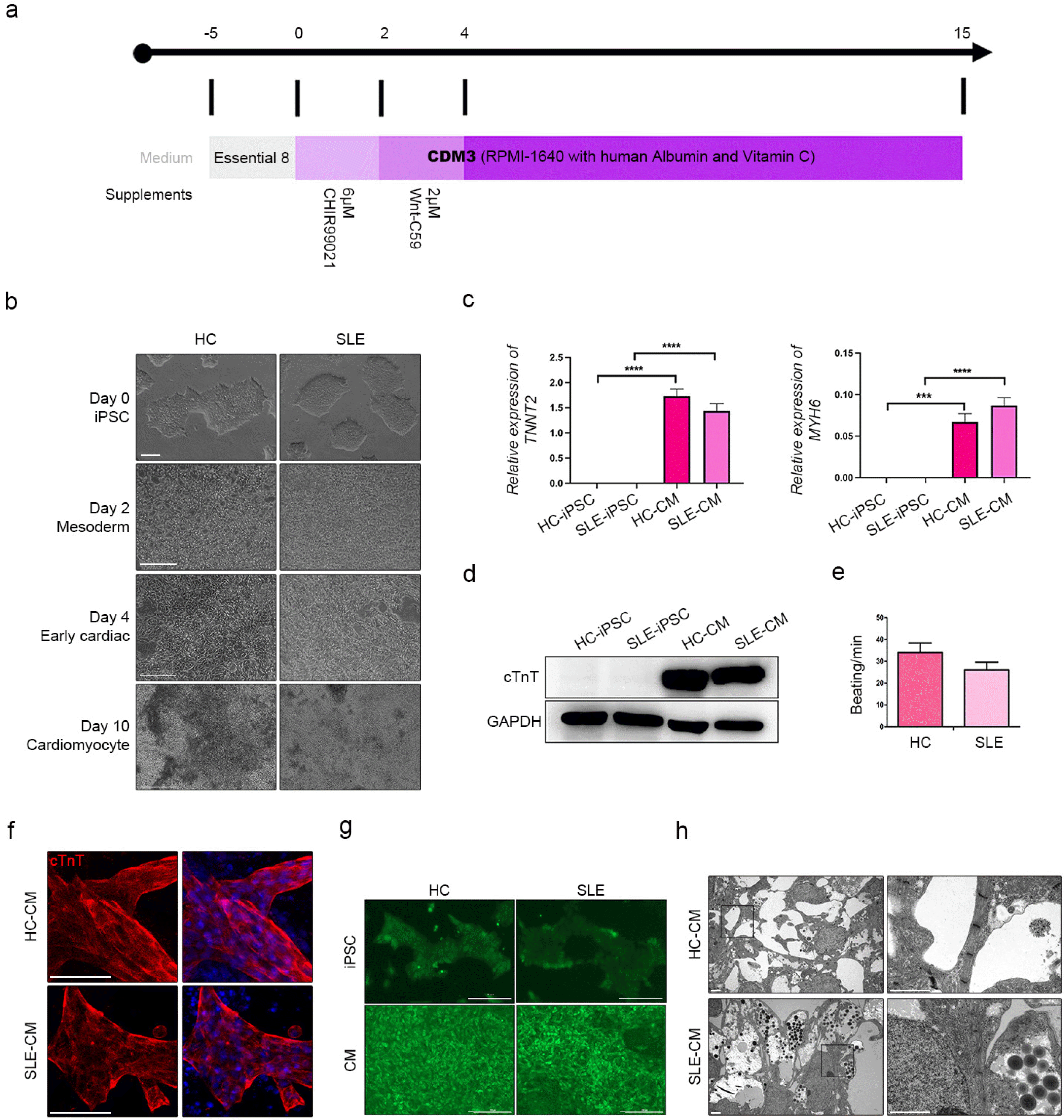
Various CM differentiation protocols have been reported for iPSCs cultured on 2D platforms (21, 22). In this study, we performed CM differentiation using small molecules. Full differentiation required 15 d (Fig. 2a). There were no morphological differences between HC-iPSCs and SLE-iPSCs in terms of CM morphology at any stage (Fig. 2b). Beating began on between day 6 and 8 of differentiation in both groups. We confirmed the expression of CM-specific markers such as cardiac troponin T type2 (TNNT2) and myosin heavy chain a-isoform (MYH6). There were no significant differences in the mRNA expression of TNNT and MYH6 among the CMs derived from SLE-iPSCs (SLE-CMs) and HC-iPSCs (HC-CMs) (Fig. 2c). On day 15 of differentiation, beating was verified for both HC-CMs and SLE-CMs (Supplementary Video S1, S2). When the protein level of the TNNT2 marker was confirmed, it was confirmed similarly to the gene expression in HC and SLE-CMs (Fig. 2d). The beating rate was slightly slower in SLE-CMs than in HC-CMs, but the difference was not significant (Fig. 2e). The cardiac muscle isoform of troponin T (cTnT) is encoded by TNNT2. Positive cTnT expression was established in HC-CMs and SLE-CMs (Fig. 2f). Calcium signals were measured to identify functional activity in the differentiated CMs (Fig. 2g). We determined that the calcium signal was weaker in the SLE-CMs than in the HC-CMs (Supplementary Video S3, S4). Filaments are representative CM structures. Whereas they were present in the HC-CMs, they were relatively absent in the SLE-CMs (Fig. 2h). In conclusion, HC-iPSCs and SLE-iPSCs both exhibited successful CM differentiation. While there were no significant differences in TNNT and MYH6 expression between SLE-CMs and HC-CMs, however, the results suggest that they might have differed structurally and functionally.
SLE patient-derived serum treatment in differentiated CMs
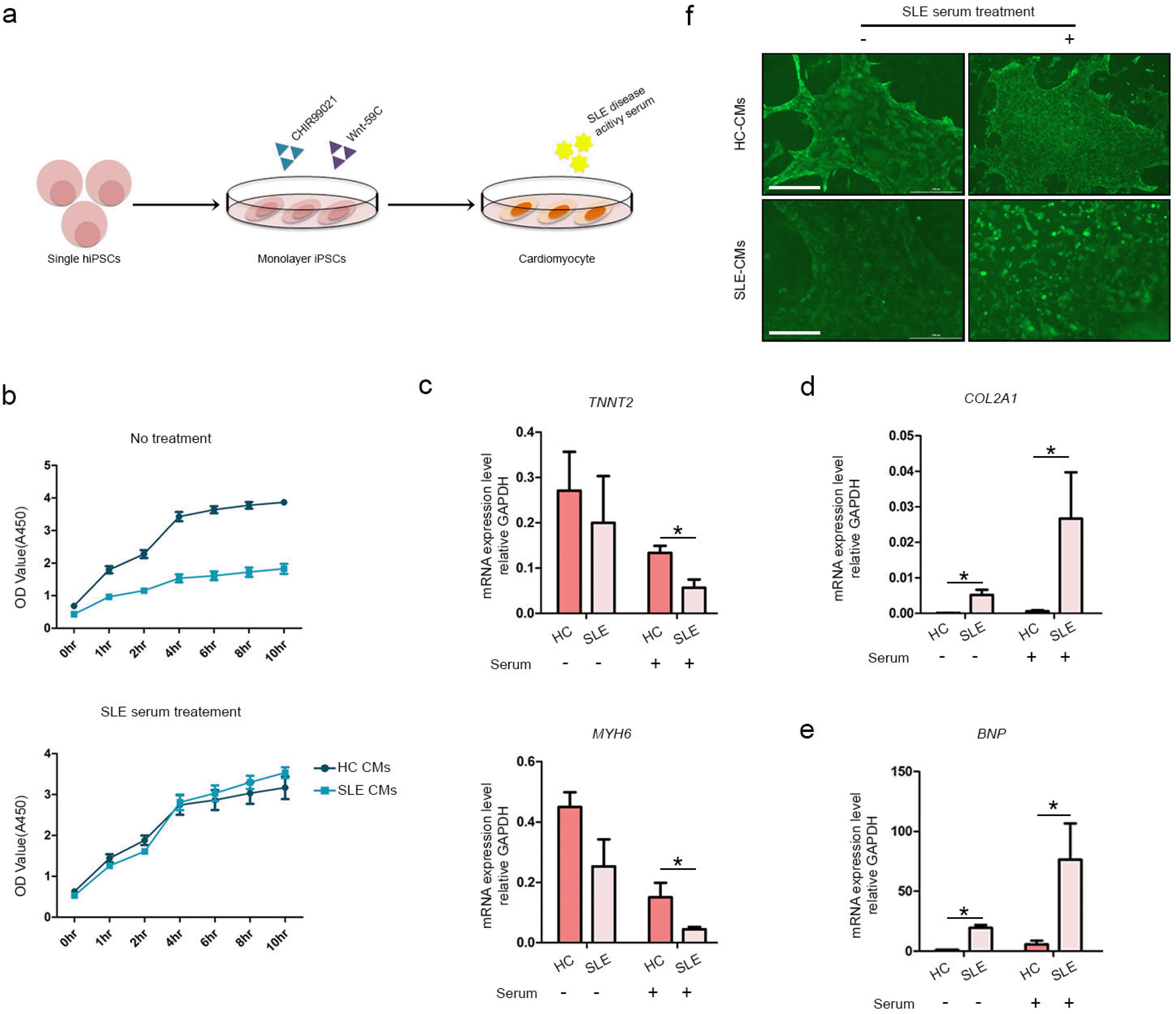
To determine the impact of the SLE environment on the differentiated CMs, we attempted to induce it by adding serum obtained from patients with SLE. We used fully differentiated iPSC-derived CMs and treated them with a mixture of SLE serum obtained from two patients and incubated for 48 hours (Fig. 3a). CCK-8 assay was performed to detect the toxicity of the serum or its impact on cell proliferation. Interestingly, SLE-CMs showed lower proliferation rate compared to the HC-CMs even in the absence of serum treatment (Fig. 3b). Treatment with SLE serum increased the proliferation rate of SLE-CMs. The serum treatment had less effect on HC-CMs. We then measured the gene expression levels in the serum-treated CMs. MYH6 and TNNT2 were significantly downregula-ted in serum-treated HC-CMs and SLE-CMs (Fig. 3c). Fibrosis and hypertrophy is suggested to increase myocardial cell proliferation (23). Fibrosis markers such as COL2A1 and hypertrophy markers such as BNP were expressed in serum-treated CMs (Fig. 3d and 3e). Both COL2A1 and BNP were upregulated in the untreated SLE-CMs compared to that of HC-CMs. SLE serum treatment further upregulated the fibrosis and hypertrophy marker expressions in both HC- and SLE-CMs; however, the expression increased in SLE-CMs. We identified calcium signaling in the serum-treated HC-CMs and SLE-CMs (Fig. 3f). Relative to untreated HC-CMs and SLE-CMs, calcium signaling was delayed in serum-treated HC-CMs and SLE-CMs (Supplementary Video S5, S6). Taken together, our results suggest that SLE-CMs show difference in cell proliferation and increased expression of fibrosis and hypertrophy-related markers even without serum treatment. Also, those properties reacted differently to serum treatment compared to that of HC-CMs. However, based on the results of the HC-CMs, the foregoing results indicated that fibrosis and hypertrophy can be induced in 2D CMs with SLE disease-activated serum in vitro. However, we were unable to maintain long-term 2D cultures as the cells detached from the culture dish and died. Hence, future research should endeavor to develop a platform that can support cell cultures over long periods of time and effectively represent chronic inflammatory disease.
3D CM differentiation using HC-iPSCs
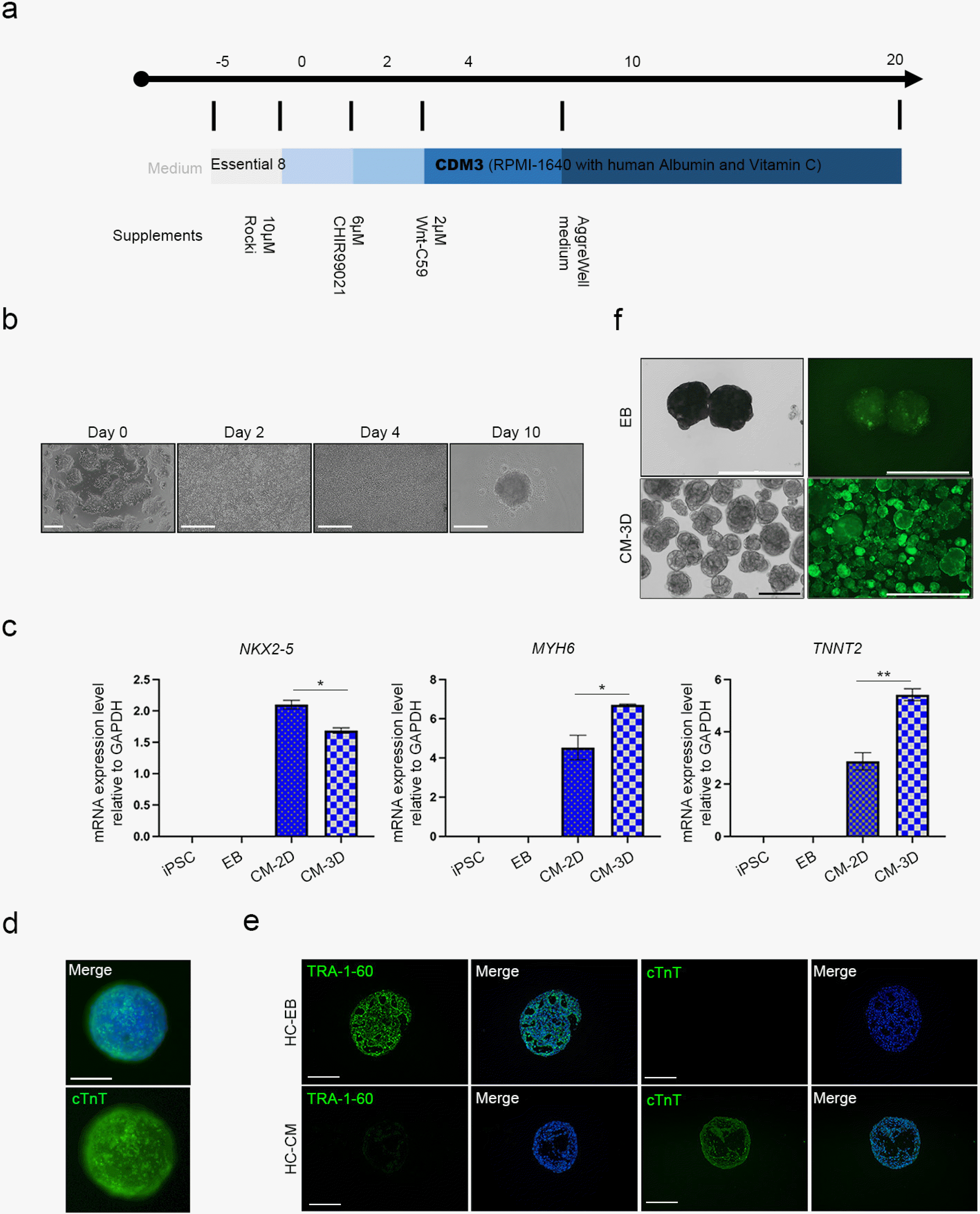
We empirically confirmed that 2D layers of CMs rapidly detach from the culture dish. Therefore, this configuration has limited applicability in disease modeling (Fig. 3). A long-term method was required to model SLE using iPSCs; therefore, we established a 3D differentiation protocol that may extend CM viability for prolonged in vitro culture (Fig. 4a). Cells were differentiated using a 2D platform until the cells started beating. Then the beating CMs were isolated into single cells with CM dissociation medium. The 3D-CMs were prepared by seeding 4×104 cells in a 96-well round plate (Fig. 4b). We then compared gene expression between 2D-CMs and 3D-CMs (Fig. 4c). The early cardiac marker NXK2-5 expression was significantly higher in 2D-CMs compared with that in 3D-CMs. However, the expression levels of the late cardiac markers MYH6 and TNNT2 were higher in the 3D-CMs than in the 2D-CMs. We then stained 3D-CMs with anti-cTnT antibody and confirmed positive cTnT expression within the CM spheroids (Fig. 4d). For further confirmation of cTnT expression, embryonic bodies (EBs) were generated by condensing the same number of iPSCs and were used as a negative control. Sections of the 3D-CMs also showed positive cTnT expression, while EBs did not show any expression (Fig. 4e). Calcium signals were also identified in the final 3D-CMs (Fig. 4f) (Supplementary Video S7, S8). Through these results, we confirmed successful differentiation of 3D-CMs, and confirmed that the 3D differentiation platform result in a more mature form of CMs which might be more suitable for SLE modeling.
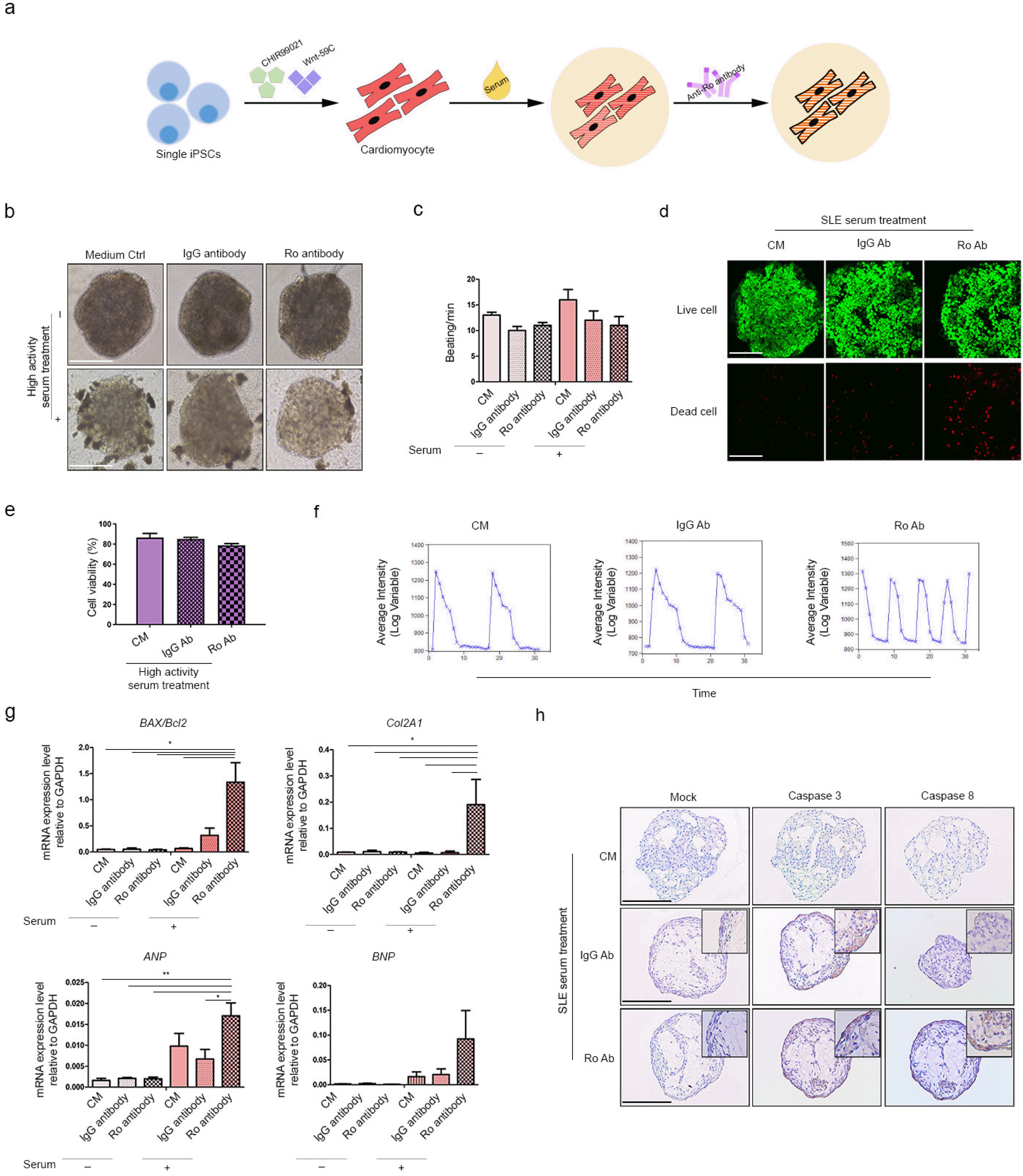
Impact of anti-Ro autoantibody on HC-CMs treated with active SLE serum
We previously demonstrated upregulation of fibrosis and hypertrophy markers in CMs treated with active SLE serum. We attempted to establish the impact of SLE serum on 3D-HC-CMs (Fig. 5a). We additionally treated anti-Ro autoantibodies as they are considered SLE biomarkers. Treatment with serum and anti-Ro autoantibodies did not alter 3D-CM morphology (Fig. 5b). There was no significant difference between groups in terms of CM beating; however, serum treatment alone seemed to slightly decrease the beating rate of CMs (Fig. 5c). The viabilities of the serum-treated and serum plus autoantibody-treated HC-CMs were 86% and 78%, respectively; it was not significantly affected by serum or autoantibody treatment (Fig. 5d and 5e). While serum alone did not affect the calcium signaling of CMs, calcium signaling became unstable in response to anti-Ro autoantibodies (Fig. 5f). We measured the expression levels of the apoptosis, fibrosis, and hypertrophy markers in the serum-treated and autoantibody-treated CMs (Fig. 5g). Apoptosis markers including the BAX/Bcl2 ratio were upregulated in response to both treatments, but anti-Ro autoantibodies treatment showed higher levels of expression. The same trends were also observed for the fibrosis and hypertrophy markers. The protein levels of apoptosis marker, caspase 3/8 was confirmed in the 3D-CMs (Fig. 5h). The expression of both caspase 3 and 8 were increased in the 3D-CMs additionally treated with anti-Ro autoantibodies. However, SLE serum treatment itself seemed to slightly increased caspase 3/8 expression, especially caspase 3. Taken all together, we confirmed that anti-Ro autoantibody treatment altered the calcium signaling of HC-CMs and further affected the expression of apoptosis, fibrosis, and hypertrophy markers in CM spheroids.
Discussion
SLE is an autoimmune disease affecting various organs by causing inflammation and tissue damage and the most common type of lupus (24). Currently, optimal stratification of SLE patients was not performed and therefore the response to available treatments was unpredictable. Universally effective and safe treatments for SLE remain unmet demands due to the heterogeneity of clinical manifestations of unpredictable reactions for current treatments (25). Though HF is associated with SLE, its pathophysiological mechanism remains unknown. Immunosu-ppressive therapies are usually administered for SLE and its cardiac complications. Several cardiac diseases are characterized by cardiomyocyte death; however, the adult heart has only a limited capacity to recover from cardiomyocyte loss or damage (26). Continuous cardiomyo-cyte loss eventually damages the heart, progressively diminishes its functionality, and leads to HF (26). Cardiac diseases tend to be silent for long periods of time, but are potentially fatal in SLE (27, 28).
End-stage organ damage in SLE is a manifestation of systemic immune dysregulation. Stem cell-based research and therapy may be valuable tools that help understand and treat this disease (29, 30). Disease modeling with iPSCs may help clarify the pathogenesis of SLE and its related symptoms. With the high pluripotency and differentiation ability, iPSCs can differentiate into the targeted cell-of-interest related to the disease. Human iPSCs can be generated from blood cells, fibroblasts, or keratinocytes. Previous studies have already reported reprogramming of SLE-patient derived iPSCs. Certain iPSCs have been generated using renal tubular cells obtained from the urine of patients with SLE as well as dermal fibroblasts and blood cells (20, 31). In this study, we generated SLE patient specific iPSCs using PBMCs and differentiated into CMs using two platforms: 1) 2D monolayer culture and 2) 3D spheroid culture using the beating cells from the 2D culture.
In previous studies involving iPSCs, disease modeling was performed using conventional 2D monolayer culture platforms (21). However, a major limitation of this system is that it cannot emulate the complex in vivo 3D environ-ment. Several studies reported that 2D iPSC differentia-tion models do not provide vital information about cell-to-cell communication. The latter depends on the biochemi-cal and biophysical support provided by the extracellular matrix (ECM) and surrounding cells characteristic of in vivo environments (22, 23, 32). Moreover, cells differentiated on 2D platforms may share traits with immature fetal cells rather than adult cells (33). In the present study, we established cardiomyocyte differentiation on both 2D and 3D platforms (Fig. 2, 4). Nkx2-5 is an early marker during differentiation of the cardiac lineage. It is usually expressed in the progenitor cells forming the cardiac crescent (34, 35). Here, we demonstrated that Nkx2-5 was downregulated whereas the mature CM markers MYH6 and TNNT2 were upregulated in the cardiomyocytes differentiated on the 3D platform (Fig. 4c). Our results corroborated those previously reported because they showed that the 3D platform generates more mature cardiomyo-cytes than the 2D platform (21). However, the 2D disease modeling platform is easy to handle and highly reproduci-ble. Hence, it might be still acceptable for modeling fetal and early-onset cardiac diseases, and the selection of the appropriate differentiation platform depending on the disease type might be critical.
As mentioned earlier, organ damage in SLE is a result caused by the dysregulated immune response. It might be possible that the CMs differentiated from HC- and SLE-iPSCs does not exhibit any major differences in morphology and cardiac marker expression; however, further analysis revealed that SLE-CMs had lower proliferation rate and increased expression levels of hypertrophy and fibrosis markers compared to the HC-CMs in 2D platform (Fig. 3). SLE serum seemed to increase the proliferation rate of SLE-CMs and amplify the genetic difference between the HC- and SLE-CMs. Therefore, it is believed that HC- and SLE-CMs do have different characters, and further examination using transcriptomic or RNA sequencing analysis might be interesting to thoroughly confirm the differences between the HC and diseased CMs.
The pathophysiology of cardiac diseases in SLE patients is far more complex. In this study, we simply tried to recapitulate myocardial toxicity that are found in SLE patients; however, there are various symptoms that can be found in SLE patients such as acute heart involvement (myocarditis) and late vascular involvement. Further development and application of a protocol that can target a specific type of cardiovascular complication may be beneficial for a more specified disease modeling of SLE.
Anti-Ro autoantibodies are associated with several autoimmune diseases including SLE and it is the most detected type of autoantibody in patients with SLE (36). Certain studies suggested a close relationship between anti-Ro antibodies and SLE onset later in life (≥50 y) (7). Conflicting data suggest a correlation between anti-Ro autoantibody titer and SLE activity (37, 38). Nevertheless, anti-Ro autoantibodies are considered critical factors in SLE research. In the present study, we subjected iPSC-derived HC-CM spheroids to anti-Ro autoantibodies that were either untreated or exposed to serum from patients with SLE. Calcium signaling patterns were altered by the treatment of anti-Ro autoantibodies (Fig. 5f). Gene expression of apoptosis, fibrosis, and hypertrophy markers were increased by the additional treatment of anti-Ro autoantibodies (Fig. 5g). Apoptosis marker expression was increased in the 3D-CMs treated with anti-Ro autoantibodies; however, the number of actual dead cells did not show significant differences between the groups. A longer observation with constant regular treatment of serum and anti-Ro autoantibodies in 3D-CMs might suggest more findings that might further connect HF in SLE with cell apoptosis in future studies.
Though our data were preliminary, they revealed that SLE patient serum and anti-Ro autoantibody treatment increased marker expression related to apoptosis, fibrosis, and hypertrophy in the differentiated CMs, which might be related to the pathogenesis in patients with SLE. Future research should endeavor to characterize the inflammatory factors in SLE patient serum and identify those that interact with anti-Ro antibody using a larger sample size.
Supplementary Materials
Supplementary data including eight videos can be found with this article online at https://doi.org/10.15283/ijsc21158.

 XML Download
XML Download