

 PDF
PDF Citation
Citation Print
Print



Introduction
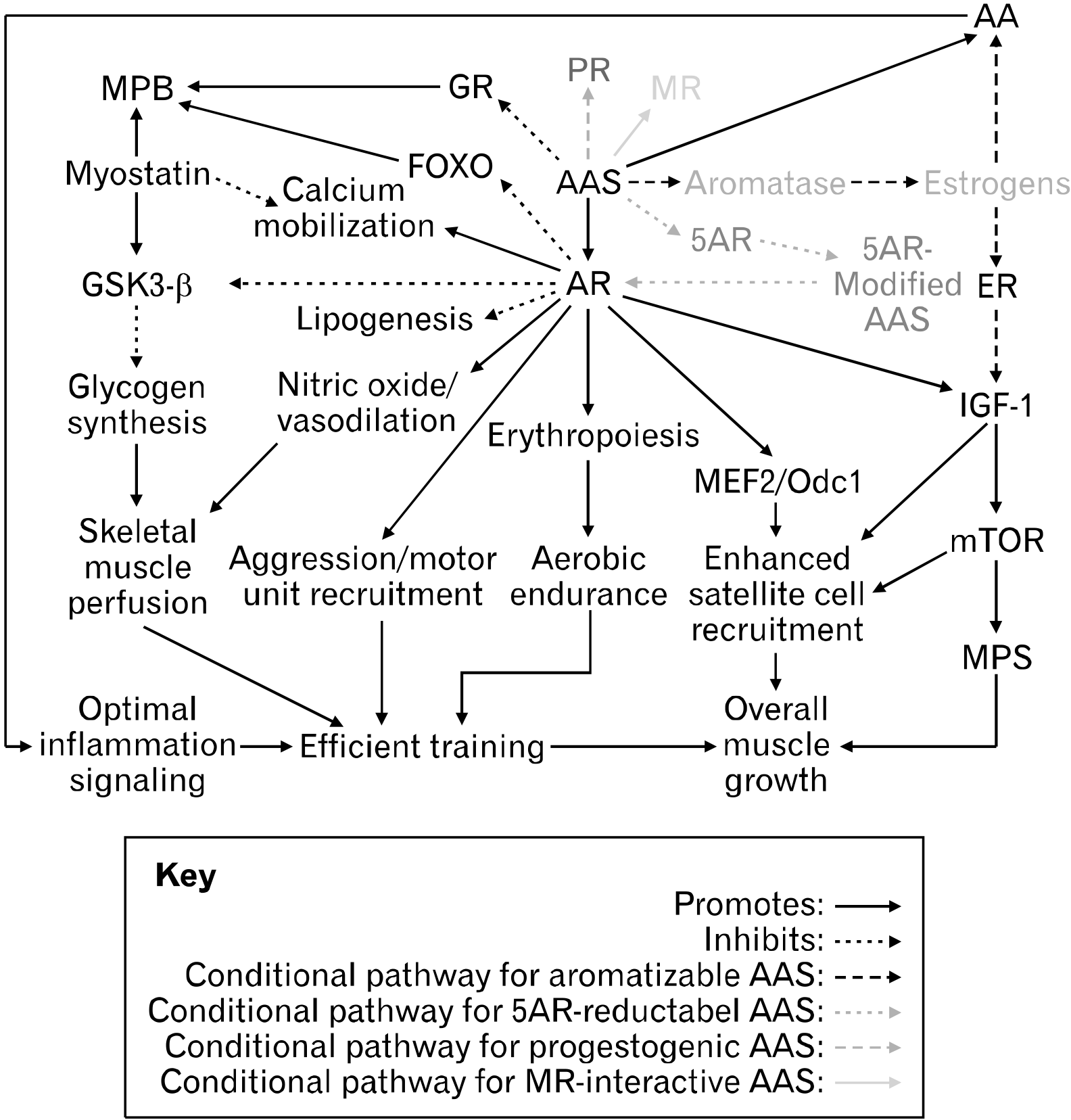
Bodybuilding is a sport in which competitors attempt to develop supreme muscularity and conditioning. At the extremes, this goal is paradoxical, as low body fat and high muscle mass are best achieved in hypocaloric and hypercaloric states, respectively1. However, one can improve their body composition to a certain extent by dedicating years of strict dieting and efficient resistance/aerobic training. Skeletal muscle growth is mediated by multiple biochemical pathways including the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), the mammalian/mechanistic target of rapamycin (mTOR), the mitogen-activated protein kinases (MAPKs), various calcium-dependent pathways, and the wingless-related integration (Wnt) protiens2. Essential hormones involved in muscle growth include insulin, insulin-like growth factor-1 (IGF-1), steroid hormones, and growth hormone (GH)3. The collaboration of these hormones and biochemical pathways results in satellite cell recruitment and a positive net protein balance following resistance exercise, ultimately yielding muscle growth over time4. Although one can gain an ample amount of muscle with training and dieting alone, it is inevitable to reach a plateau in muscle gains. This is largely attributed to myostatin, a potent negative regulator of skeletal muscle mass involved in muscle homeostasis5. Myostatin inhibits many of the previously mentioned muscle growth pathways, significantly blunting satellite cell recruitment and muscle protein synthesis (MPS)5-7. Presuming the most optimal conditions for anabolism (i.e., top-tier genetics and near-perfect training/diet/recovery), a fat-free mass index (FFMI) of about 25 appears to be the upper limit of muscle mass for drug-free (natural) male bodybuilders6.
FFMI=(Lean weight in kg/2.2)/{(Feet×12.0+Inches)×0.0254}2
For instance, the average healthy man has an FFMI of ≤19, while a ≤178 cm bodybuilder who weighs ≤88 kg at 10% body fat would have a FFMI of ≤25 using the equation above. Many bodybuilders are notorious for pursuing every competitive edge. Despite potential moral and health consequences, some decide to take performance-enhancing drugs (PEDs) to drastically improve their body composition. Since the 1930s, a variety of PEDs have been developed, allowing athletes to surpass their natural limits, feasibly attaining FFMIs of over 306. This reinforces the ability of PEDs to grant users body compositions and athletic performance achievements far beyond physiological capabilities. Importantly, there is an extensive range of PEDs in use today, as the term “PED” describes a drug with performance-enhancing benefits in any metric. Though, considering the focus of this review, the term “PED” will refer to drugs with myotropic capabilities. As such, this review will outline the mechanisms of anabolic-androgenic steroids (AAS) and selective androgen receptor modulators (SARMs) since these drugs are often regarded as foundational PEDs of modern bodybuilding. In addition, myostatin inhibitors will also be described as these drugs appear to be a promising new generation of PEDs. Users of PEDs include both athletes and nonathletes, as well as those with a competitive agenda and those with body dysmorphia. It is estimated that between 2.9 and 4 million Americans have used AAS in their lifetime, including a significant portion of adolescent users8. AAS use, at any age, may result in short- and/or long-term side effects, such as permanent stunted growth in adolescent users, while all male AAS users risk anabolic steroid-induced hypogonadism (ASIH) (Section: Anabolic-Androgenic Steroid-Induced Hypogonadism)7,8. Despite an extensive body of literature on the mechanisms of muscle hypertrophy4 and a comparably minuscule, but still significant scope of research on PEDs, many prospective/current PED users lack a full comprehension of either. This lack of comprehension appears to be a prominent limiting factor for both bodybuilders and clinicians alike in making sensible decisions for their own/patient’s health. The present review provides readers with information that may aid in the development of an efficient PED protocol, characterized by abundant benefits and minimal adverse effects. There are various organizations, such as the World Anti-Doping Agency (WADA), to deter the use of PEDs in athletics, suggesting a generally negative public perception, or perhaps even fear, of PEDs. By understanding how these compounds affect the body and endogenous growth pathways, one may realize that PEDs are not inherently hazardous, although risks are inevitable with misuse. PEDs may be able to pose several benefits when administered under proper protocols, especially in the clinical setting of muscle- and bone-wasting diseases. In this respect, the purpose of the present review is to describe how the implementation of AAS, SARMs, and myostatin inhibitors enhance muscle growth by manipulating endogenous growth pathways and briefly elucidate tips for practical application.
Endogenous Testosterone Production
Physiological and behavioral sexual dimorphisms arise from considerably different hormone profiles between men and women, particularly in the sex steroids, androgens and estrogens9. Numerous steroidogenic stimuli, as well as intratesticular factors, play a role in the intricate regulatory network of testosterone synthesis. Natural synthesis of testosterone in adult males occurs primarily in the Leydig cells of the testes which produce over 95% of the total circulating testosterone in men10. This route of testosterone synthesis is mediated by the hypothalamic-pituitary-gonadal (HPG) axis8. In this endocrine circuit, the hypothalamus releases gonadotropin-releasing hormone (GnRH), which stimulates the anterior pituitary gland to release luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH and FSH stimulate the testicles to produce testosterone and sperm, respectively. In the Leydig cell plasma membrane, LH initiates a cascade of events involving the LH receptor (LHR), G proteins, adenylate cyclase, and cyclic adenosine monophosphate11. Cytochrome P450 (CYP) enzymes catalyze the transformation of cholesterol into bioactive steroids. First, cholesterol is converted into pregnenolone, which is then converted into progesterone by 3-β-hydroxysteroid dehydrogenase (3-β-HSD) enzymes12. Another CYP enzyme converts progesterone into androstenedione, which can be finally metabolized into testosterone by type III 17-β-HSD12. The adrenal cortex contributes to the rather trivial remaining portion of testosterone in men by secreting precursors such as dehydroepiandrosterone and androstenedione that can be metabolized into testosterone in peripheral tissues10. Without exceptional environmental factors/stresses, endogenous testosterone production follows a diurnal pattern, usually being highest in the morning and lowest in the evening13. The normal range of serum testosterone concentration in men is 300 to 1,000 ng/dL while peak testosterone levels consistently under 300 ng/dL is medically considered male hypogonadism13. As hydrophobic molecules in a mostly hydrophilic internal body environment, endogenous steroid hormones utilize transport proteins to stabilize their bioavailability in circulation. Only about 2% of total testosterone is unbound to proteins and can freely diffuse into cells14. This proportion of the total testosterone is called free testosterone and is the principal bioactive form of testosterone15. The remaining testosterone in circulation is bound to either sex hormone-binding globulin (SHBG) or albumin. Compared to SHBG, albumin is significantly more prevalent throughout the body but with an inversely proportional binding affinity for sex hormones14.
Anabolic-Androgenic Steroid-Induced Hypogonadism
The HPG axis is regulated by a negative feedback system, meaning that high levels of testosterone and other androgens, as well as estrogens, inhibit endogenous testosterone/sperm production, while low testosterone/androgen levels are com-pensated by increased GnRH/LH secretion12. As such, AAS suppress endogenous testosterone and sperm production8. Upon ceasing AAS use, the serum concentration of gonadotropins and endogenous testosterone production may remain suppressed for an indefinite period of time, sometimes for many years or even for life depending on the dose/type/duration of AAS used and the individual. The medical term for this state is ASIH16. Due to its abiding nature, ASIH is probably the greatest risk associated with AAS use in men. The implementation of post-cycle therapy drugs, such as human chorionic gonadotropin and antiestrogens (i.e., clomiphene citrate and tamoxifen) are often utilized following an AAS cycle to accelerate the recovery of endogenous testosterone and fertility16. However, full recovery is never guaranteed, and many long-term AAS-users risk permanent ASIH which can only be ameliorated by hormone replacement therapy8. Despite this risk and the many adverse effects associated with AAS use, some athletes still choose to take AAS, perhaps due to their undeniable performance and physique-enhancing benefits.
Androgens Enhance Muscle Growth
In 1935, European researchers artificially synthesized testosterone and noted its ability to enhance athleticism7. Within a few decades, the use of testosterone as a PED became popularized in competitive athletics, and today, the use of AAS underlies the sport of bodybuilding15. Among the many AAS used, testosterone appears to be the best understood demonstrated by the various studies outlining its effects9,17,18. Essentially, all AAS are analogs of testosterone and ligands for the androgen receptor (AR) and so, many of the anabolic effects of testosterone reported in literature can be extrapolated to describe the broad nature of AAS, at least in relation to the AR. In reality, there are still unique properties associated with each AAS (Section: The Divisions of Anabolic-Androgenic Steroids). Many of the anabolic processes such as localized acute inflammation, IGF-1 production, MAPK integration, phosphoinositide 3-kinase (P13K)/Akt/mTOR/70S6K activation, and satellite cell recruitment are enhanced by androgens like testosterone (Fig. 1). Testosterone has shown to upregulate GH as well as both circulating and intramuscular IGF-14,14. To support the subsequent elevation in MPS, it has been suggested that some androgens sensitize satellite cells to growth factors19. Crosstalk between forkhead box o (FOXO) atrogenes and Akt is also modulated by androgens and influences net protein balance20. Augmentation of glycogen synthesis, insulin sensitivity, metabolic rate, and nutrient sensing/uptake as well as inhibitory effects on lipogenesis by testosterone administration have also been reported20. These effects optimize nutrient partitioning and facilitate skeletal muscle perfusion during exercise, further promoting muscle growth. Notably, androgen treatment has shown to enhance myonuclear accretion as shown by the fact that the number of myotubes with more than five myonuclei approximately doubled in those incubated in testosterone compared to control21. This is perhaps due to testosterone stimulating the production of IGF-1 precursors and upregulating myogenic regulatory factors (MRFs) such as myogenin (MyoG), myoblast determination protein 1 (MyoD), and paired box 7 (PAX7)22,23. Moreover, the greater myogenic commitment/fusion index of satellite cells associated with testosterone treatment is attributed to androgen-mediated phosphorylation of extracellular signal-related kinase (ERK)-1/2 and enhanced Wnt/frizzled receptor protein signaling22,24. Varying effects of androgens on myostatin and its antagonist, follistatin, expression have been reported. Dubois et al.25 reported that androgens promote myostatin signaling, likely to restrain their own anabolic actions. However, in the murine atrophy model, androgen resupplementation increased muscle-specific expression of follistatin, and in another study, testosterone administration reduced myostatin in previously atrophied cells, but not in normal or previously hypertrophied cells21,26. These findings may appear to be contradictory. However, the uncertainty may be settled when considering the anabolic/catabolic reversible reaction model and the set point of muscle mass presented in this review (Sections: The Anabolic/Catabolic Reversible Reaction Model and The Set Point of Muscle Mass). In this respect, the anabolic actions of androgens promote skeletal muscle hypertrophy until a compensatory rise in myostatin deters excessive deviation from the set point of muscle mass.
Androgen Receptor Structure and Expression
Many of the myotropic effects of androgens are mediated through the AR. Resistance training upregulates AR content in humans4. Sinha-Hikim et al.27 have shown that testosterone treatment increases AR density in cultured muscle satellite cells. As expected, men have significantly higher levels of AR messenger RNA (mRNA) and a greater AR activation response to endogenous testosterone compared to women28. The AR is a member of the type I nuclear receptor family which consists of additional steroid hormone receptors such as the estrogen receptor (ER), the progesterone receptor (PR), the glucocorticoid receptor (GR), and the mineralocorticoid receptor (MR)29. These steroid receptors share a common structure consisting of a highly variable amino-terminal domain, also referred to as the A/B domain or N-terminal domain; a central conserved DNA-binding domain (DBD), also referred to as the C domain; a hinge region, also referred to as the D domain; and a carboxy-terminal ligand-binding domain (LBD), also referred to as the E domain30. The DBD remains concealed until receptor activation and contains two zinc fingers with high cysteine affinity suitable with their function in attaching the steroid receptor to DNA. Ligand-receptor binding is facilitated through a hydrophobic ligand-binding pocket (LBP) composed of several αβ-helices in the LBD. This LBP is highly flexible to accommodate a variety of ligands and serves as the cardinal interface between ligand and receptor31. Phosphorylation of the hinge region, which is between the DBD and LBD, facilitates nuclear localization of the receptor32. AR activity is mediated through the constitutive (ligand-independent) activation function (AF)-1 in the N-terminus33. AF-1 is the target of various growth factors that activate the AR ligand independently. Unlike AF-1, AF-2 of the LBD is ligand-dependent and essential in full activation of the receptor32. Both AF-1 and AF-2 influence the recruitment of coactivators/corepressors and modulate the AR’s conformation and interaction with DNA. Furthermore, the ligand-dependent crosstalk between the N-terminus and C-terminus stabilizes the interaction between ligand and receptor30. The coactivator interaction surface of the AR is in the AF-1, and this is a distinguishing feature of the AR among the type I nuclear receptors32. The AR is expressed widely throughout the body, including the cardiovascular, reproductive, immune, and musculoskeletal systems9. Both undifferentiated satellite cells and myocytes express ARs, and testosterone promotes the commitment of pluripotent mesenchymal cells into the myogenic lineage21,27. Therefore, one may infer that the AR content in existing myofibers as well as stem cells influence the myotropic responses to androgens.
Androgen Receptor Activation
In the absence of a ligand, the AR is bound to heat shock proteins (HSPs) that maintain the stability of the receptor28. Free testosterone binds to ARs located in the sarcolemma, cytosol, or nucleus of mature muscle fibers. Both genomic and no-genomic actions are involved in the myotropic effect of androgens, however, it is agreed that genomic AR activation and subsequent gene transcription is crucial for maximizing androgen-induced hypertrophy24,34. Nonetheless, rapid nongenomic effects of androgens, such as calcium influx, MAPK/ERK activation, and nitric oxide release, overlap with and likely support/modulate the genomic effects of the AR9. In the classical/canonical (genomic) pathway of androgen action, activation of the AR leads to its dissociation from HSPs, homodimerization, conformational changes (i.e., exposure of the nuclear localization sequence), nuclear translocation, coregulator recruitment, and gene transcription. In the nucleus, zinc fingers of the DBD bind to DNA sites called hormone response elements29. Specifically, ARs bind to androgen response elements to modulate gene transcription and protein synthesis. This involves the recruitment of histone acetyltransferase enzymes and essential coregulators which facilitate the binding of the TATA-binding protein28. The subsequent recruitment of general transcription factors (i.e., RNA polymerase II) initiates the transcription of androgen target genes35. Evidently, the genomic androgen/AR pathway is highly dependent on the ligand stability/binding affinity, AR content/conformation, and coregulator recruitment.
Androgen Receptor Target Genes
Most of the tangible effects of androgens on skeletal muscle are a result of gene transcription and protein translation. For instance, the myostatin gene is highly responsive to androgen signaling and there appears to be crosstalk between the AR, β-catenin, follistatin, and other transforming growth factor β (TGF-β) members in the regulation of myogenic differentiation25,34. Androgen-responsive targets such as FOXO and mTOR are also components of the classical AR vector (Fig. 1)20,36. The positive effects testosterone has on glycogen synthesis, insulin sensitivity, and nutrient partitioning can be attributed to its ability to modulate genes such as growth factor receptor bound protein 10 (GRB10), Phk-γ, and Lipin 120. Wyce et al.37 conducted the first genomewide mapping of AR binding in skeletal muscle cells. Researchers identified over 30,000 potential AR-binding sites in the human genome, among which were genes for protein degradation (i.e., muscle atrophy F-box protein [MAFbx]), various microRNAs (miRNAs) involved in satellite cell proliferation/differentiation, as well as the AR gene itself and myocyte enhancer factor 2 (MEF2)-related genes37. Though MEF2 transcription factors do not possess myogenic activity alone, the interaction of MEF2 with MRFs yields synergistic activation of muscle-specific genes crucial for myogenic differentiation38. Wyce et al.37 also found that androgen treatment increased the expression of sarcomere integrity and muscle contraction genes as well as miRNAs involved in myoblast differentiation. Rana et al.36 conducted a similar study on AR target genes, in which they concluded that in males, androgen/AR signaling promotes peak muscle mass by prolonging the proliferation period of myoblasts, allowing the formation of additional myoblasts prior to differentiation and fusion. This effect appears to be mediated via the AR target gene, ornithine decarboxylase 1 (Odc1)39. Other genomic actions of androgens in various models include increased AR expression and calcium mobilization (calcineurin/nuclear factor of activated T cells [CnA/NFAT] signaling), ERK1/2 activation, polyamine biosynthesis, nitric oxide release/vasodilation, glycogen synthase kinase 3-β (GSK3-β) inhibition, and phosphorylation of the P13K/Akt/mTOR pathway28,36. Considering the thousands of possible combinations of AR-binding sites in the human genome and potential ligand-receptor complex configurations, the distinct effects of different AAS likely arise from their unique androgen signaling targets (i.e., one AAS may preferentially induce Odc1 but not Lipin 1, while another AAS may have reciprocal effects). Targeting specific genes, as opposed to specific receptors, emerges as another promising field to expand the scope of PED research (Section: YK-11 and Obstacles to Myostatin Blockade). Besides gene-specificity, the distinguishing features of AAS regarding anabolic/catabolic pathways arise, at least in part, from their interaction with endogenous enzymes.
Dihydrotestosterone and 5-α-Reductase
Testosterone can be transformed into various active and/or inactive compounds by endogenous enzymes15. Some enzymes, such as those found in the liver, can metabolize testosterone into less potent androgens such as androstenedione. On the other hand, 5-α-reductase (5AR) irreversibly transforms testosterone into a more potent androgen called dihydrotestosterone (DHT) by reducing the double bond between the fourth and fifth carbon atoms15. Compared to testosterone, DHT has a higher binding affinity for the AR and dissociates three to four times slower from the AR upon binding31,40. However, DHT can be further metabolized by 3-α-HSD into the biologically inactive compound, 3-α-androstanediol7. Thus, the activity of testosterone largely depends on the presence of tissue-specific enzymes. Skeletal muscle tissue generally maintains very low 5AR activity but high 3-α-HSD activity7. Therefore, in the absence of synthetic AR ligands, the primary AR ligand within skeletal muscle is testosterone itself41. In areas with high 5AR activity, such as in bone, lung, brain, adipose, and male reproductive tissue, DHT effectively competes testosterone for AR binding42. During pubertal development, DHT is largely responsible for the growth and development of the male reproductive organs. Consequently, in adult males, unregulated testosterone administration can lead to adversities such as an enlarged prostate, acne, and balding of the scalp as these tissues generally express high AR density and 5AR content43.
Estrogen and Aromatase
Testosterone, as well as any androgen with a Δ-4-3-keto configuration, can be aromatized into 17-β-estradiol and/or other estrogens by aromatase44. Since aromatase activity is significant in adipose tissue, individuals with higher body fat may experience worse estrogenic side effects in response to the administration of Δ-4-3-keto AAS14,45. Nonetheless, estrogens and ERs of various tissues maintain a wide variety of physiological functions. Favorable metabolic effects (i.e., insulin sensitivity) and glucose/lipid homeostasis are mediated by ER-α and ER-β, respectively46. In steers, estradiol administration alone has shown to increase IGF-1 mRNA expression, satellite cell proliferation, as well as mTOR and MAPK/ERK signaling, suggesting that the anabolic effects of testosterone are at least partially mediated by its conversion into estrogens46. In L6 and C2C12 cells, estrone treatment increased myoblast growth, while both estrone and estradiol treatment increased c-Fos expression47. Estrogens have also been proposed to enhance AR activity by increasing AR density and binding affinity14. Some AAS, particularly aromatizable AAS, have demonstrated the ability to remodel the plasma lipidome and increase arachidonic acid (AA) which is likely associated with estrogen signaling48,49. AA is a crucial component of the acute inflammatory signal following resistance training that kickstarts downstream anabolic processes50. Therefore, bodybuilders need adequate estrogen, though estrogen levels should be tightly controlled, especially shortly before a bodybuilding competition. This is because excessive estrogen is associated with gynecomastia, fluid retention, adiposity, and toxicity51,52. On a bodybuilding stage, subcutaneous fluid and fat retention take away from the athlete’s conditioning, while obvious gynecomastia hinders the overall aesthetic of the physique. To combat these adverse effects, bodybuilders usually deploy antiestrogens, such as selective ER modulators and aromatase inhibitors, alongside aromatizable AAS8. It is important not to completely eradicate estrogen with these drugs for prolonged periods of time since estrogens maintain optimal lipid profiles, bone mineral density, and brain function45,49.
The Divisions of Anabolic-Androgenic Steroids
Testosterone, although a powerful anabolic, lacks optimal tissue-selectivity, especially at supratherapeutic doses32. Other endogenous androgens have similar limitations. Thus, a variety of AAS have been developed with the goal of dissociating beneficial anabolic effects from unwanted adverse ones. A detailed and comprehensive encyclopedia on PEDs has been written by Llewellyn14 titled William Llewellyn’s Anabolics. In this book, Llewellyn14 describes all AAS as either bioidentical/derived versions of testosterone, nandrolone, and/or DHT and in this sense, three divisions of AAS emerge: testosterone/testosterone derivatives, DHT/DHT derivatives, and nandrolone/nandrolone derivatives. AAS of the same division are often, but not always, united by similar endogenous enzyme/cellular milieu interactions, non-classical steroid receptor binding patterns, and AR-binding affinities (Sections: The Anabolic/Androgenic Ratio and Steroid Cross-Reactivity). Importantly, AAS still agonize the AR while maintaining their own unique characteristics53. For instance, some testosterone/testosterone derivatives are noted for their ability to convert into estrogens and DHT/DHT equivalents. Members of this AAS division often serve as a testosterone “test” base in an AAS stack. In theory, test bases mimic the physiological balance of testosterone, estrogens, and DHT, which would otherwise be suppressed by other AAS. An optimal balance of this trio helps to maintain mood, libido, and overall health17. In this regard, the use of testosterone is likely the most ideal test base, though the optimal dose may depend on the type/dose of other PEDs being used. Moreover, utilizing a low dose “testosterone only” cycle as one’s first experience with AAS may be pertinent as testosterone is bioidentical, generally well-tolerated at lower doses, and readily converted into physiological androgens and estrogens. DHT derivatives are marked by their unique reduced double bond between the fourth and fifth carbon atoms53. Like their parent hormone, DHT derivatives bind strongly to the AR and are often regarded as the most purely anabolic and tissue-selective family of AAS since most have little-to-no aromatase/5AR interactions and some members of this family contain antiestrogenic properties14. However, this also conveys that DHT derivatives generally possess milder overall anabolic effects in comparison to aromatizable AAS since estrogen is essential for maximal muscle growth. AAS that bind strongly to the AR sometimes have concomitant binding affinities for SHBG40. In this regard, the DHT derivative, mesterolone, although a relatively weak anabolic agent alone, can be used to confine SHBG and enhance the bioavailability of other AAS better suited for anabolism. The final division of AAS is the nandrolone family. Nandrolone is also referred to as 19-nortestosterone as it is generated by substituting the methyl group on the 19th carbon of testosterone with a hydrogen molecule14. Derivatives of nandrolone contain the same substitution and are collectively known as “19-nors” in the bodybuilding community. The unique structural modification of 19-nors decreases steric hindrance and subsequently increases AR-binding affinity54. In skeletal muscle, nandrolone binds to and activates the AR rather unaltered since this milieu lacks 5AR. Nandrolone appears to be less virilizing than testosterone and has been historically used preferentially over testosterone to treat osteoporosis in women14,17. The improved tissue-selectivity of nandrolone can be largely attributed to its interaction with 5AR in nonskeletal muscle regions. Whereas the 5AR metabolism of testosterone generates a more potent androgen, the reduction of nandrolone by 5AR in androgenic tissues (i.e., in the scalp and prostate) yields a weaker androgen, 5-dihydro-19-nortestosterone. Thus, nandrolone may be less likely to cause androgenic alopecia and benign prostatic hyperplasia (BPH) compared to testosterone17. Unfortunately, these beneficial characteristics of 19-nors are paired with notable side effects. 19-nors do not provide sufficient estrogen/DHT via enzymatic conversion17,43. As such, a test base is recommended when using nandrolone/nandrolone derivatives to prevent side effects such as sexual dysfunction and neurotoxicity. 19-nors also appear to suppress endogenous testosterone production to a greater degree than other divisions of AAS. Accordingly, Pomara et al.55 reported the ability of nandrolone to downregulate CYP enzymes involved in steroidogenesis. Also, despite little-to-no aromatization, some 19-nor users report gynecomastia14. These unique adverse effects of 19-nors may stem from their ability to bind to and activate the PR, which is speculated to have synergistic effects with ER, aromatase, and estrogens14,56. One option may be to incorporate antiprogestins (i.e., cabergoline) to prevent these side effects. However, androgens with progestonic activity may be more capable of myogenesis compared to exclusively androgenic/estrogenic compounds since both female sex hormones, estrogens and progesterone, are important regulators of muscle protein turnover57. In human endometrial stromal cells, 17-β-estradiol plus progesterone, but not 17-β-estradiol or progesterone alone, induces follistatin-related gene expression58. At any rate, additional patterns of steroid cross-reactivity are prevalent with most AAS and greatly influence their specific effects.
Steroid Cross-Reactivity
All steroid hormones resemble a similar core structure derived from cholesterol and as a result, the structures of all type I nuclear receptors are also very similar, allowing multiple steroid ligands to bind and agonize/antagonize their activity29,51,59. As shown by the testosterone/cortisol ratio in the model of overtraining, androgens and glucocorticoids generally antagonize each other’s actions by displacing one another from their respective receptors4,59,60. Appropriately, some of the protein-sparing effects of AAS are caused by GR antagonism19,52. Various interactions between AAS and the ER have been described. DHT-derived androgens, drostanolone and mesterolone, have shown to possess antiestrogenic activity14. Takeda et al.61 found that methyltrienolone (methylated trenbolone) acts as a potent antagonist of the MR, perhaps explaining the “dry-appearing” physique associated with trenbolone use. Beyond 19-nors, progestin activity has been reported with methandienone, a testosterone-derived AAS62. Therefore, in addition to AR-binding affinity and enzymatic conversion (or lack thereof), one must evaluate AAS based on their potential interactions with multiple steroid receptors. For instance, it would be sensible to deploy antiestrogenic/DHT-derived compounds (i.e., drostanolone and oxandrolone) in the few weeks prior to a bodybuilding competition to yield a dry and lean appearing physique. On the other hand, water-retentive/estrogenic AAS (i.e., nandrolone and testosterone) would prove useful during a bulking phase to lubricate joints and facilitate skeletal muscle perfusion which would complement increased training load/volume as well as long-term myofibrillar hypertrophy.
The Anabolic/Androgenic Ratio
The anabolic/androgenic (A/A) ratio is a rough estimate of how myogenic vs. virilizing one AAS is relative to another. The A/A ratio can be calculated by using bioassays representing anabolic (i.e., levator ani) and androgenic (i.e., seminal vesicles) tissues in castrated male rats53. Following AAS administration and a period of time to allow for hypertrophy, the A/A ratio is determined by dividing the weight gain of the anabolic tissue by the weight gain of androgenic tissue. The Hershberger bioassay is the most well-known A/A ratio calculation method53,63. Nonetheless, the A/A ratio of an AAS is extrapolated to represent its general anabolic and androgenic effects compared to a reference steroid. Testosterone is the most commonly used reference steroid with a widely accepted A/A ratio of 1:1, although methyltestosterone and nandrolone sometimes serve as references for oral and 19-nor AAS, respectively14,64. Appropriately, the A/A ratio of an AAS is highly dependent on its AR-binding affinity and interaction with endogenous enzymes like 5AR. In general, nandrolone-derived and DHT-derived AAS tend to maintain a higher A/A ratio than testosterone-derived AAS63,64. However, it would be unreasonable to rank the three divisions of AAS from “best” to “worst” based only on their A/A ratio since there is significant variability in the A/A ratio even among members of the same division59,63,64. The A/A ratio also fails to account for the interaction of AAS with enzymes other than 5AR as well as potential synergistic applications (i.e., mesterolone) and steroid cross-reactivity. Furthermore, the tissues used to calculate the A/A ratio are not accurately representative of skeletal muscle tissue. The levator ani, although low in 5AR activity, contains ten times the AR concentration of skeletal muscle53. This would likely result in an overestimation of the anabolic effects of an AAS on peripheral skeletal muscle tissue. Though it may seem that a higher A/A ratio is always more favorable, there are considerable limitations to solely relying on the A/A ratio when assessing the overall value of an AAS.
17-α-Alkylation and 17-β-Esterification
Oral administration of raw testosterone results in its rapid degradation into inactive compounds via hepatic first-pass metabolism. One the other hand, parenteral administration (i.e., injections) of raw testosterone results in a brief peak in hormone concentration, followed by rapid clearance before substantial anabolic effects occur7. The 17th carbon of AAS is the primary target to overcome these physiological barriers. 17-α-alkylation involves the addition of a methyl or ethyl group to the 17th carbon of an AAS, allowing a significant portion of the drug to survive degradation during hepatic first-pass metabolism7. For some oral AAS, biotransformation preceding systemic exposure may be necessary in producing desired effects as seen with oxymetholone and its main bioactive metabolite, mestanolone65. Examples of methyl-based 17-α-alkylated AAS include the orally available steroids, methyltestosterone, oxymetholone, oxandrolone, and stanozolol. Although 17-α-alkylation may enhance oral bioavailability, this very modification is also hepatotoxic52,53. High doses/prolonged use of 17-α-alkylated steroids are associated with liver carcinoma, cholestatic jaundice, and impaired excretion function42. To mitigate liver damage, it would be sensible for enhanced bodybuilders to rely mainly on injectable AAS and sparingly implement oral AAS. 17-β-esterification involves the attachment of an ester to the 17-hydroxy group of the AAS7. As the ester is gradually hydrolyzed, the active steroid is released into the bloodstream over an extended period allowing for a substantial window of anabolic activity53. As such, the half-life of an AAS is positively correlated to its ester length.
Selective Androgen Receptor Modulators
Despite the development of AAS that exhibit superior tissue-selectivity in comparison to testosterone, AAS are still associated with an extensive list of side effects such as dangerously elevated hematocrit levels, dyslipidemia, liver/kidney damage, and ASIH42. In an attempt to ameliorate these drawbacks, researchers developed a new class of AR-dependent anabolic drugs called SARMs66. Notably, any ligand with a specific modulating effect on the AR is clinically considered as a SARM and this includes agonistic, antagonistic, steroidal, and nonsteroidal AR ligands. Therefore, all AAS as well as the antiandrogen, bicalutamide, are technically SARMs. However, the novel class of PEDs we are referring to by the term “SARMs” are nonsteroidal and have anabolic effects on bone/muscle tissue. As a result of their polarity, SARMs generally possess good oral bioavailability while being minimally hepatotoxic, higher AR-binding affinity than most AAS, and appear to interfere with the endogenous testosterone production to a lesser degree than AAS as SARMs are less likely to cross the blood-brain barrier (BBB) and interrupt the HPG axis31,32. Some current SARMs include analogs of aryl propionamide, bicyclic hydantoin, quinoline, and tetrahydroquinoline which either agonize or antagonize the AR depending on the tissue53.
The Tissue-Specificity of Selective Androgen Receptor Modulator
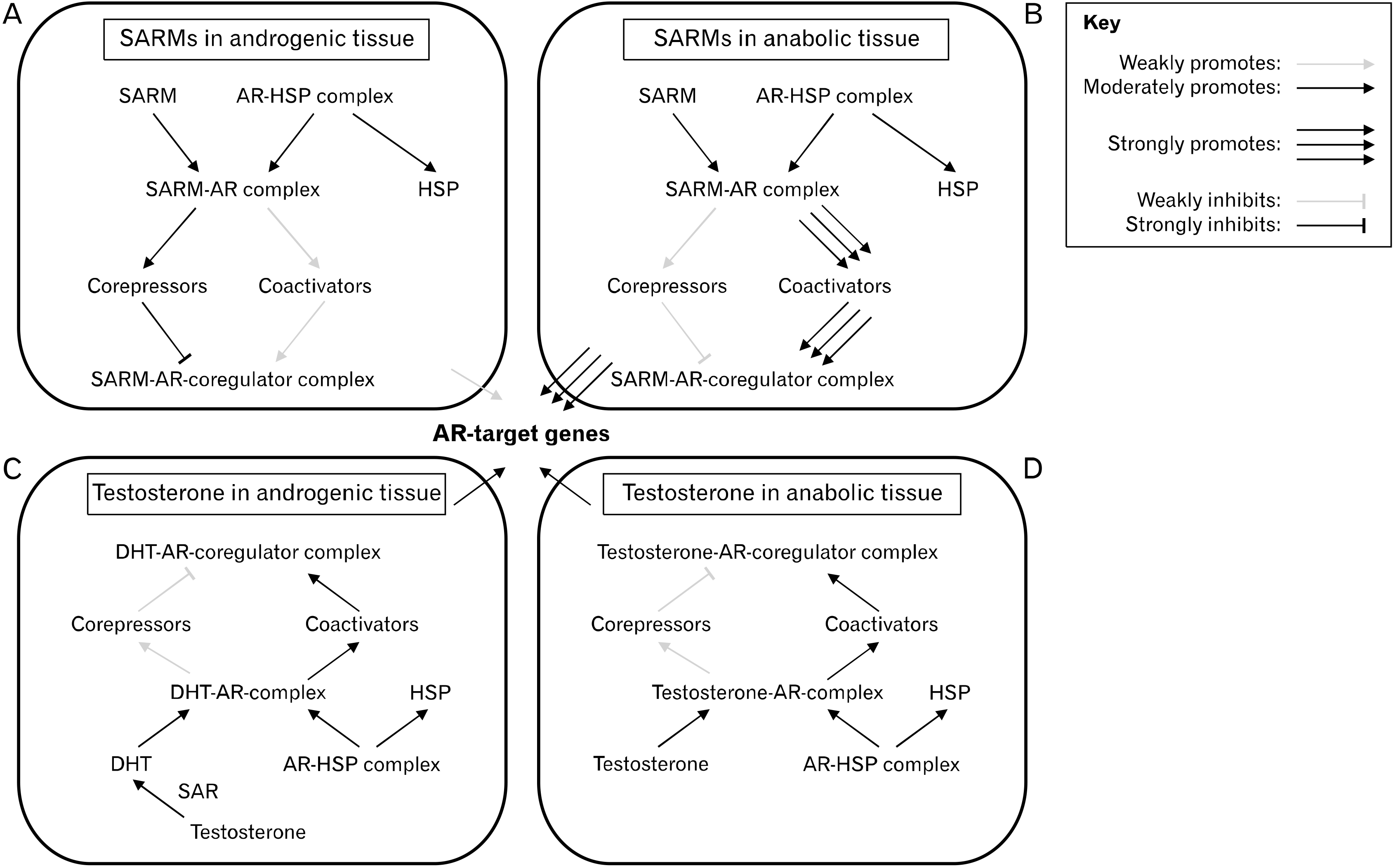
One SARM, RAD-140, has been reported to have an A/A ratio of around 90:1 as it acts as a partial antagonist in prostate tissue but as a full agonist in skeletal muscle tissue67. The exact mechanism by which SARMs exert their tissue-selective effects has yet to be determined, although it is likely related to their unique AR binding and coregulator/transcription factor recruitment as well as their lack of aromatization/5AR-reduction53. The magnitude of AR activation is highly dependent on whether the binding ligand promotes or inhibits the interactions between the two functional (N-terminal and C-terminal) domains of the AR30. The conformation of the resulting N/C interaction (or lack thereof) affects which coregulators and transcription factors are recruited, thereby affecting how/if the entire complex interacts with DNA33,35. When AAS bind to the AR, the hydrophobic residues of the LBP accommodate the steroid core, while the hydrophilic amino acids of the LBP form hydrogen bonds with the polar atoms of AAS31. These interactions affect the stability, specificity, and selectivity of ligand binding and thus, may explain some of the distinct phenotypical characteristics among various AAS. However, AAS are limited by their rigid steroid plane, which hinders the structural flexibility of the ligand-receptor complex and narrows the range of customizability in terms of selective coregulators/transcription factors and/or DNA binding conformation66. On the other hand, the more pliable nonsteroidal SARMs can be designed to induce specific conformational changes to the LBD, promote/inhibit N/C terminal interaction, modulate surface topology and thermodynamic partitioning, alter protein-protein interactions between the AR and coregulators/transcription factors, and ultimately yield tissue-specific gene regulation68,69. In the prostate-specific antigen (PSA) enhancer site of LNCaP cells, DHT recruited steroid receptor coactivator 1 (SRC-1) but not nuclear receptor corepressor (N-CoR), acting as an overall strong agonist while an aryl propionamide-derived SARM recruited both SRC-1 and N-CoR in these cells, acting as an overall weak agonist in this androgenic tissue model32,33,67. Conversely, in skeletal muscle tissue, the SARM preferentially recruited coactivators over corepressors and acted as an overall strong agonist32. These findings have led to the coactivator hypothesis which describes testosterone-bound ARs and SARM-bound ARs to recruit distinct profiles of coregulator proteins (Fig. 2)69. Of course, the tissue-specific coregulator profile may also affect the A/A ratio of SARMs and selective coregulator recruitment may depend on extrinsic factors (i.e., inflammation). The latter is demonstrated by bicalutamide, which differentially recruits coregulators based on the presence of interleukin-832. In summary, it appears that each SARM results in a unique AR-ligand conformation depending on its structure and the cellular milieu, subsequently affecting coregulator recruitment, DNA interaction, and target gene expression.
Balancing Selective Androgen Receptor Modulators and Anabolic-Androgenic Steroids
One SARM under clinical development, LGD-4033, has shown to increase lean body mass of healthy men (n = 76; age, 21–50 years; body mass index, 18–32 kg/m2) by an average of 1.21 kg in 21 days at a dose of 1 mg/day as part of a placebo-controlled study70. No significant changes in hemoglobin, PSA, liver enzymes, or QT interval were noted, but a dose-dependent decrease in high-density lipoprotein and endogenous testosterone production was observed. Therefore, it appears current SARMs still lack true tissue-selectivity and since many bodybuilders are willing to use PED doses that are 10-fold the dose used in clinical studies, additional undocumented adverse effects may arise8. SARMs have incredibly high binding affinities for the AR, but with respect to the diminishing returns of AAS and the anabolic/catabolic reversible reaction model, this also conveys that their slope of diminishing returns is probably much steeper (Section: The Anabolic/Catabolic Reversible Reaction Model). As such, high doses of SARMs generally yield subpar muscle gains compared to high doses of AAS14. This is probably because some of the off-target effects of AAS support the direct anabolic actions of AR activation while SARMs are specifically designed to be as targeted as possible. Estrogen conversion, erythropoiesis, increased aggression, and GR antagonism associated with AAS are some off-target effects that may prove to be useful if leveraged properly (Fig. 1). For instance, higher androgen levels in the brain are associated with aggression, which may aid in achieving maximum training intensity, and an increased hematocrit from androgen signaling in bone marrow may yield aerobic endurance benefits7,53. AAS users also commonly experience enhanced neuromuscular coordination and motor unit recruitment, and this postulated to be a result of androgens increasing neurotransmitter synthesis, upregulating neurotransmitter expression, and accelerating synaptic vessel cycling71. However, each of these benefits are paired to caveats such as increased risk of violent behavior, hypertension, and neurotoxicity52,72. Therefore, neither AAS nor SARMs are superior: each class of PEDs maintains its unique benefit/side effect profile. Nonsteroidal SARMs appear to err on the side of caution with generally milder adverse effects but also milder anabolic benefits, while AAS exhibit higher risk, but also higher reward. It may be pertinent to combine a test base with certain SARMs and/or AAS to achieve synergistic anabolic effects or to neutralize adverse effects. For instance, one may pair RAD-140 with an AAS that stimulates prostate enlargement so that the risk of BPH is comparatively neutralized, while both compounds would exert myotropic effects (Fig. 2). The wide range of tailored functional characteristics of SARMs suggests the possibility of developing a compound able to ameliorate ASIH when using suppressive androgens. This ideal SARM would be characterized as orally bioavailable (for convenience), have customized interaction with the BBB, and be selectively antagonistic/neutral of the AR in key components of the HPG axis while agonizing the AR in skeletal muscle tissue. Additional PEDs, such as insulin and GH, exert their effects beyond the AR73,74. As expected, these drugs also possess unique benefits and risks, however, they will not be detailed in this review.
Myostatin Inhibitors
To date, no single SARM or AAS has completely isolated the anabolic effects from the androgenic effects75. Even with a completely tissue-selective AR-dependent PED, it is logical to assume that myostatin, among other catabolic factors, will increase in parallel to muscle gains, resulting in diminishing returns and a subsequent ceiling of muscle mass. As shown in the upcoming anabolic/catabolic reversible reaction model, maximum muscle growth would only be achieved by combining pro-anabolic agents with equally potent anticatabolic agents. Endogenous myostatin inhibitors, such as follistatin and myostatin propeptides, regulate the activity of myostatin. However, at physiological concentrations, these endogenous myostatin inhibitors fail to block the concurrent increases in myostatin with increasing muscle mass76-79. In normal mice, two injections of soluble type II activin receptor B (ActRIIB)-Fc, a decoy receptor for myostatin and similar TGF-β members, induced up to a 60% increase in skeletal muscle mass in just 2 weeks80. Thus, the inhibition of myostatin may emerge as one of the most effective strategies to increase muscle mass. Some myostatin inhibitors such as YK-11 elicit their myotropic effects by upregulating the expression of follistatin81.
YK-11 and Obstacles to Myostatin Blockade
YK-11 is a steroidal gene-selective partial agonist of the AR, or in other words, a SARM82. Inhibiting myostatin by targeting the AR is possible because both myostatin and follistatin are subject to androgen signaling25,34,79. In C2C12 myoblast cells, additional key anabolic targets of the AR such as MyoG, Mfy5, and MyoD are upregulated with the administration of YK-1183. Myogenic effects of YK-11 also involve NF-κB signaling as it has been suggested to attenuate muscle wasting during sepsis84. Unlike full AR agonists, YK-11 prevents N/C interaction, and it has been suggested that YK-11 may also interfere with the ability of other AR ligands (i.e., DHT) to induce conformational changes to the AR necessary for full activation85. This characteristic poses a potential limitation when combining YK-11 with a wide range of PEDs because AAS and other SARMs highly depend on full AR agonism to exert their myotropic effects18,24,66. Furthermore, there are numerous other barriers to myostatin inhibitors. Gum bleeding, telangiectasia, and erythema following soluble ActRIIB administration has hindered its clinical progression35,86. Myostatin inhibitors that function via follistatin upregulation also have their own drawbacks. Follistatin inhibits pituitary FSH release, and thus, artificially upregulating follistatin harms fertility87. With respect to safety and efficacy, YK-11 has yet to be tested in human clinical trials. However, there are certainly a significant number of athletes who have used the PED, demonstrated by the development of drug tests for YK-11 in athletic competitions81. Myostatin inhibition, at least via follistatin upregulation, may also increase the risk of injury in athletes. Tendons of myostatin knockout rats have 20% less peak strain compared to wild-type controls88. Similar treatments in mice caused comparable effects, resulting in tendons that were small, brittle, and hypocellular89. Follistatin knockout mice exhibit craniofacial and rib defects90. As such, TGF-β superfamily members appear to regulate the balance between stem cells and tissue homeostasis beyond skeletal muscle tissue5,91. With respect to resistance exercise adaptations, myostatin may serve to ensure tendon integrity to match the increasing tensile load generated by growing contractile protein tissue. Consequently, one should practice extra caution when utilizing myostatin inhibitors due to their potential to increase injury risk. Further research may reveal novel insight on the dichotomous tissue-specific nature of TGF-β expression in response to androgen signaling. For instance, androgens like DHT tend to promote facial hair growth but simultaneously induce the miniaturization of scalp hair follicles in those prone to androgenic alopecia92. Perhaps all AR ligands, AAS included, maintain gene-selective effects depending on the tissue type and corresponding unique cellular milieus composing distinct coregulator profiles and epigenomes. This may further explain differing physiological effects of various androgens and suggests the possibility of engineering new AR ligands through a gene- and/or epigene-selective approach. Nonetheless, myostatin inhibition as a whole remains another relatively untapped field for novel anabolic agents.
The Natural Bodybuilder’s Dilemma and Diminishing Returns of Performance-Enhancing Drugs
Due to the extensive list of physiological limitations that natural bodybuilders face, it has become obvious that one can concurrently choose only up to two of the following three: extreme muscularity, extreme conditioning (low body fat), or PED abstinence. Even in the case where one decides to stay drug-free and maximize either leanness or size, they will likely never achieve the same level of development in either metric as they otherwise would using PEDs that amplify their efforts. Hence, these athletes ultimately face what we have coined the natural bodybuilder’s dilemma; stay natural and face an inevitable stagnation in physique improvement, or take PEDs, that is, become enhanced, and continue to make progress beyond natural limits. The latter option seems to promise more reward in the bodybuilding niche as shown by the fact that, unlike most athletic competitions, the highest caliber bodybuilding leagues usually do not drug test for PEDs, such as the National Physique Committee and International Federation of Bodybuilding and Fitness (IFBB). A natural bodybuilder choosing to go down the enhanced route might expect a one-to-one ratio between the dose of PEDs and muscle gains acquired, suggesting the possibility of literally unlimited muscle mass. Yet, there appears to be a blatant diminishing returns effect with most PEDs. A study on the dose-response relationship of testosterone by Bhasin et al.93 demonstrated that the amount of lean body mass acquired per unit of testosterone declines nonlinearly at higher doses. These diminishing returns are probably, at least in part, due to increasing basal myostatin levels as Dalbo et al.79 found a concomitant upregulation of mature myostatin protein levels alongside the increase in skeletal muscle mass and enhanced myogenic lineage in a rodent model after the administration of AAS. Thus, even with pharmaceutical support, it is practically guaranteed that one will experience subsequent plateaus. The occurrence of muscle gain plateaus can be explained by the set point of muscle mass and the anabolic/catabolic reversible reaction model which are explained below sections.
The Set Point of Muscle Mass
Enhanced bodybuilders can manipulate their biochemistry, namely their hormones and even their genes, on demand, while natural bodybuilders remain susceptible to many unfavorable limitations. However, enhanced bodybuilders are still vulnerable to some physiological limitations, like myostatin, unless these hurdles are specifically targeted (i.e., myostatin inhibition). There appears to be a link between the limits of muscle growth and the set point theory. This theory describes the tendency of body weight to remain stable, especially in periods of energy restriction, due to various homeostatic mechanisms (i.e., altered energy expenditure and/or changes in ghrelin/leptin secretion)94,95. In tandem, the widely accepted physical stress theory (PST) distinguishes the varying thresholds of physical stress that induce tissue atrophy, maintenance, hypertrophy, or death96. In skeletal muscle tissue, proper doses of mechanical and biochemical stresses result in anabolic adaptations. According to the PST, to continue gaining muscle, one will have to expose their body to increasing stress demands while concurrently ensuring adequate recovery. It is apparent that humans have finite rates of nutrient digestion, absorption, and incorporation, as well as a ceiling for training intensity, frequency, and volume. In this regard, skeletal muscle growth occurs until the body reaches what we call the set point of muscle mass. This set point of muscle mass is directly correspondent to the level of anabolic stimuli and nutrients, and once it has been reached, muscle growth stagnates due to homeostatic regulators such as myostatin or simply, insufficient anabolic stimuli to cause a further adaptive response. Thus, muscle growth can be thought of as “closing the gap” to reach equilibrium/steady state between the current state of muscularity and the target set point of muscle mass based on current and forthcoming anabolic stimuli/nutrient intake. Like the diffusion of water, the larger the gradient (i.e., the difference between the current state and the target state of muscularity), the faster the adaptations generally occur given recovery is also proportional. Hence, this is why adaptations happen most rapidly when one first exposes themselves to a stimulus and exponentially slows down until a new stimulus is given. Along the lines of the original set point theory, there seems to be a tendency to retain physiological levels of acquired muscle mass as shown by the maintenance of strength, myofiber size, and muscle mass with only one-third the exercise volume used to initially obtain these metrics97. This is supported by the PST as the threshold of stimulus for tissue maintenance is substantially lower than the threshold of stimulus for tissue hypertrophy96. Skeletal muscle is a plastic tissue capable of fiber recompositing, however, the retention of myonuclei and sustained epigenetic modifications described as the muscle memory phenomenon support the propensity of muscle size to remain stable despite a rather dynamic fiber composition98,99. To reach a new set point of muscle mass, one must adjust their anabolic stimuli and/or nutrient intake accordingly. For instance, if the current FFMI of a male bodybuilder is 23 and the target set point of muscle mass based on his anabolic stimuli/nutrient intake is an FFMI of 25, he will gradually accrete muscle mass until the FFMI of 25 is reached so long as the anabolic stimuli/nutrient intake remain stable. If this bodybuilder decided to start implementing 250 mg of testosterone per week along with a 10% increase in calorie intake and 20% increase in training volume, his new target set point of muscle mass may be an FFMI of 26 or 27, and consequently, he would grow muscle tissue until reaching this new target set point of muscle mass.
The Anabolic/Catabolic Reversible Reaction Model
A stable muscle protein turnover indicates equilibrium between MPS and muscle protein breakdown. Hence, the widely accepted FFMI natural limit of 25 appears to be the steady state/equilibrium point, in other words, the set point of muscle mass, for genetically gifted healthy men who have maximized all the natural anabolic stimuli and perfected their diet (nutrient intake). For both enhanced and natural athletes, we have found it comprehensible to visualize muscle protein turnover and anabolic/catabolic stimuli as a reversible reaction model that emulates a basic chemical reaction. This is illustrated by the following anabolic/catabolic reversible reaction model:
Nutrients+Anabolic stimuli↔Muscle growth+Compensatory muscle growth inhibitors
Nutrients can serve as anabolic stimuli (i.e., amino acids activate mTOR), but require additional anabolic stimuli, such as growth factors and muscle damage, to assemble into functional muscle proteins. Meal distribution is also an important factor to consider, thus by the term “nutrients”, we mean the consumption, as well as the processing efficiency of nutrients (i.e., bioavailability and tissue uptake). A lack of nutrients, insufficient anabolic stimuli, and/or excessive muscle growth inhibitors would pull the reaction to the left and result in muscle loss. However, one must also consider muscle memory, which would likely delay this process to a degree. When one accretes new muscle tissue, the reaction is pulled to the right and the compensatory muscle growth inhibitors (i.e., myostatin) increases concomitantly. To continuously shift the reaction to the right, or in other words, to keep progressing, one must evoke more potent anabolic stimuli. Reciprocally, without continuously increasing anabolic stimuli and/or nutrients, one will hit an eventually reach equilibrium, or in other words, a plateau. PEDs are essentially artificial anabolic stimuli that allow one to surpass their natural ceiling of lean mass, forcefully pulling the reaction to the far right. However, most PEDs still result in an increase of compensatory muscle growth inhibitors as shown in the studies by Bhasin et al.93 and Dalbo et al.79 and therefore, another inevitable stagnation in muscle growth occurs. Upon reaching this plateau, rather than simply increasing the dose of PEDs, one should first consider how to mitigate limiting variables. This is because PEDs often pose higher health risks at higher doses41,52. For instance, an enhanced bodybuilder with an attenuated acute inflammatory response to resistance exercise due to depleted ω-6 stores would probably benefit more from supplementing AA as opposed to simply increasing the dose of PEDs50. Thus, an optimal PED protocol should entail one to recognize limiting factors, garner as much muscle growth from the lowest efficacious dose and understand the unique effects of candidate ergogenic aids so that the overall desired outcomes outweigh the adverse effects. With respect to the set point of muscle mass and considering the health risks of PEDs, one should ensure that all physiological variables, such as training efficiency, sleep quality, and nutrient intake, are optimized before manipulating PED-related variables to surpass a plateau. Only once all relevant physiological variables have been perfected should one consider the implementation of PEDs. One should also keep in mind the potential benefits of intentional plateaus, such as scheduled deload weeks and periodizing intensity/volume/technique to peak the body for a competition100. Enhanced bodybuilders often rotate between periods of high PED dose intake and low PED dose intake (this is known as blasting and cruising) and frequently change the type of PEDs used throughout the year14. In theory, this allows some time for the body to heal from potential damage caused by PED use while refreshing and re-sensitizing skeletal muscles to the anabolic effects of PEDs and potentially attenuating year-round myostatin elevation. Therefore, like training periodization, intentionally implementing short plateaus or even periods of slight regression may be beneficial in the long run when it comes to PED use. When surpassing natural limits with PEDs, one must also consider the accumulation of organ stress (i.e., increased load on the heart, kidneys, and liver) with greater muscle mass, regardless of how that new muscle is accreted. Thus, even with advanced pharmacology, there is still an absolute limit to muscle growth and developing an efficient PED stack requires comprehension of the basic mechanism of action of common PEDs.
Conclusions
When attempting to truly maximize one’s physique, one must refrain from oversimplifying nutrition, training, and recovery and consider every possible variable/limiting factor. This process becomes exponentially more complex when pursuing competitive bodybuilding since the implementation of PEDs and conditioning become additional factors to consider. There are numerous internal mechanisms to deter extreme muscle growth and prevent excessive deviation from homeostasis. The use of PEDs may allow one to surpass their natural limit, but often exhibit blatant diminishing returns and come with an array of side effects. Perhaps the question to ask as a bodybuilder would be not how to get as muscular as possible, rather, it would be more practical to ponder the most efficient means to achieve a top-tier physique. In this sense, the most efficient approach to PED use would be to first master the variables of natural training and then to add one variable at a time, starting with the safest/most well-tolerated PEDs (i.e., testosterone) and gradually advancing to higher doses and alter-native compounds to match the increasing demand of anabolic stimuli necessary for continuous progress. Of course, in doing so, one should consult with their doctor, get regular health tests, and maintain an otherwise healthy lifestyle. Additional research on PEDs will not only minimize health complications among bodybuilders, but also serve clinical importance as drugs that spare muscle and bone tissue are highly valuable in treating cachexic diseases. Nevertheless, bodybuilding is as much a science as it is a sport and a form of art.
 XML Download
XML Download