

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cerebral amyloid angiopathy (CAA), which is a disorder involving intracranial microangiopathological lesion, is the main cause of secondary subarachnoid hemorrhage (SAH) in the elderly37). The main pathological feature of CAA is the deposition of amyloid-like substances in the small vessel walls of the pia mater and cortex1). Dementia, psychiatric symptoms, transient neurological dysfunction, and recurrent and/or multiple cerebral lobar hemorrhage (CLH) are the clinical features of this disorder.
CAA mostly occurs in the elderly, while familial CAA has an earlier age of onset. The incidence and severity of CAA increase with age23). There is no difference between the incidence rates in men and women22). CAA was first reported in the last century39). However, the relationship between CAA and cerebral hemorrhage was only seriously examined in the 1970s. CAA accounts for approximately 2% of the overall causes of cerebral hemorrhage and 38% to 74% of the causes of nontraumatic nonhypertensive intracerebral hemorrhage in the elderly4).
Multifocal CLH in the cerebral cortex and subcortical white matter of elderly patients is an indication that CAA might be present. CAA was once considered a rare pathological condition, but it has recently attracted attention as an important cause of nontraumatic nonhypertensive CLH in the elderly17272835). However, the in vivo diagnosis of CAA and CAA-related cerebral hemorrhage remains difficult to make, which causes challenges in its treatment35612151824). In this study, we performed a retrospective analysis of 10 cases of CAA-related CLH that were treated and then confirmed by autopsy in our hospital. Our objective was to provide information that could improve the ability to make in vivo diagnoses of CAA.
MATERIALS AND METHODS
Subjects
The clinical specimens included 245 brain autopsy samples that were collected at the Department of Neurology of our hospital from January 1983 to January 2013. All of the cases of CAA-related CLH were confirmed with hematoxylin and eosin and Congo red staining. Ten cases of CAA-related CLH were selected for this study.
Research methods
The clinical data, including information about age, sex, clinical manifestations, bleeding sites, and causes of death, of the 10 cases of CAA-related CLH were retrospectively reviewed. Following autopsy, the brain tissue samples were fixed in 10% formalin for more than 3 weeks and subsequently sliced into 10-µm-thick horizontal or coronal sections. The brain slices were taken from the bilateral frontal, parietal, temporal, and occipital lobes, cerebellum, and shell core. The screened slices were then stained with hematoxylin and eosin, Congo red, and Bodian's silver and examined with light microscopy and polarized light microscopy.
RESULTS
Clinical data
The 10 cases of CAA-related CLH included six men and four women who were 55 to 78 years old, with a mean age of 63.5 years. Among these, two presented with a single CLH, and eight presented with multifocal CLH, which is defined as two or more separated CLHs. In addition to amyloidosis and β-amyloid (Aβ) staining in the vessel walls of small and medium-sized vessels in the subarachnoid space, leptomeninx, and cortex, there was a scattered distribution of senile plaques (SPs), amyloid cores, and Aβ staining in the cortex. The 10 cases met the CLH-related Boston diagnostic criteria for CAA16). The small vessels around the bleeding lesions showed positive Congo red staining and no other pathological changes. All of the CAA cases in the study also exhibited a SAH and had lesions primarily in the small arteries of the cerebral cortex and deep meningeal blood vessels. These lesions included microaneurysms, double barrel-like changes, multifocal arteriolar clusters, obliterative onion skin-like intimal changes, fibrinoid necrosis of vessels, neurofibrillary tangles (NFTs), and SPs.
Six patients were over 60 year old, and two were over 70 years old. Two cases had a history of hypertension, and two had a history of diabetes. Four patients smoked, two consumed alcohol, and none had a history of drug addiction, traumatic brain injury, or any other encephalopathy. None of the patients had a systemic bleeding tendency or was taking anticoagulant or antiplatelet medications. The duration from CLH onset to death in the 10 cases ranged from 1 day to 22 days. CLH presented with the following symptoms : headache (5 cases), limb paralysis (8), convulsions (6), and coma (9), including three cases of immediate coma. Lumbar punctures were performed in six cases after the onset of CLH, and they all showed bloody cerebrospinal fluid (CSF) with increased protein levels (Table 1).
Pathologic observations
The brain specimens of the 10 cases weighed 1325 g to 1610 g, with 16 fresh and four old lobar hemorrhage lesions. The hematoma bled into the subarachnoid space in all 10 cases and into the ventricle in one case.
Microscopic observations
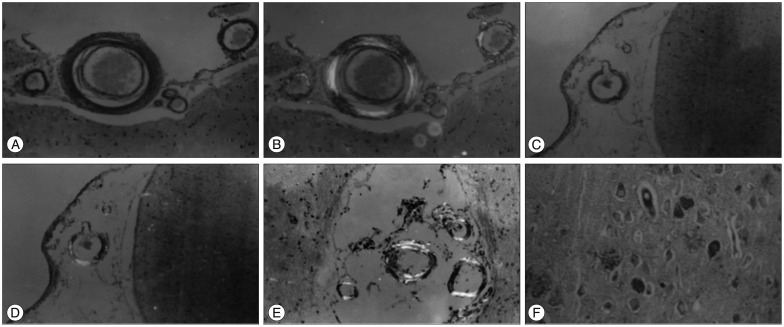
Microscopic observations revealed that the main vessels showing CAA lesions were located around the occipital lobe, temporal bone, parietal cortex, meninges, and subcortical white matter. Amyloid-like deposits were found in the arterial tunica media and tunica adventitia. Samples from all 10 cases showed positive Congo red staining, and yellowish-green birefringence was seen under polarized light, mainly in the small blood vessel walls (Fig. 1A, B). Different degrees of NFTs were observed in eight of the 10 cases, SPs were observed in five cases, and two exhibited granular degeneration.
CAA-associated diseases were also observed in the 10 cases. There were two cases with microaneurysms (Fig. 1C, D), four with double-barrel-like changes, five with multiple small arterial plexus changes (Fig. 1E), four with occluded onion-like endometrial changes, and seven with blood vascular-type cellulose wall necrosis.
Silver staining of the brain-intraparenchymal NFTs was visible in a few slices; the nerve fibers appeared degenerated, thickened, and fused; SPs were also observed (Fig. 1F).
DISCUSSION
Definition and pathogenesis of CAA
CAA is usually divided into sporadic CAA and familial CAA, which is an autosomal dominant disorder. CAA lesions mainly occur in the small arteries and capillary walls of the cerebral cortex, subcortex, and leptomeninx, and they largely exhibit a limited and irregular distribution. The cerebral veins are usually not involved. The etiology of sporadic CAA remains unclear. CAA is caused by the deposition of insoluble Aβ peptides and other amyloid-like substances in the leptomeningeal and cortical arteries, arterioles, and capillary walls, which results in certain clinical and pathological features2931). Different theories have been suggested for the origin of intracerebral Aβ. The systemic theory states that Aβ that is in the circulating blood is deposited in the cerebral blood vessels, which results in blood vessel weakening and collapse of the blood-brain barrier30). The second theory suggests that amyloid protein, which forms Aβ, is generated by the supportive cells that connect the perivascular microglial cells and cerebral blood vessels2). The third states that brain cells produce Aβ. In the normal state, Aβ is constantly degraded and removed and thus only exists in the brain parenchyma in the soluble form and in trace amounts. When the balance of Aβ metabolism is destroyed, Aβ is deposited among cells as a highly insoluble protein. During this process, SPs are formed by the protrusions of degenerated nerve cells and reactive glial cells, and Aβ is deposited onto the vascular walls to cause CAA.
Clinical manifestations and epidemiology
Given increasing life expectancies and the effective control of hypertension, CAA is common. The incidence of CLH has exhibited an increasing trend. In fact, CLH is the most common clinical manifestation of CAA, and approximately 30% of the cases with CLH limited to one lobe are caused by CAA. CAA-related CLH usually involves more than one lobe, and survivors of a first hemorrhage commonly experience recurrence. The predilection sites of CAA-related CLH remain controversial. The bleeding sites in CAA are often reported in the occipital lobe, rear temporal lobe, or superficial areas of the frontal lobe13). Other reports have indicated that the frontal (posterior part of the forehead) and parietal lobes are the most common sites, the temporal and occipital lobes are less affected, and the cerebellum is rarely involved10).
The symptoms of CAA-related CLH include sudden onset of headache; neurological symptoms, such as limb weakness, sensory loss, and visual or language changes (symptoms depend on the lobe involved); decreased consciousness; nausea; and vomiting36). Given that CAA-related CLH is often accompanied by subarachnoid hemorrhage (SAH), cervical rigidity and positive meningeal irritation are usually observed in the examination, and the CSF that is obtained with lumbar puncture consists primarily of a uniform bloody substance. Spotted bleeding, which is light bleeding involving many small vessels, may cause recurrence or transient neurological symptoms, or it may rapidly develop into dementia. In this study, the 10 cases of CAA-related CLH presented with the following clinical symptoms : five cases of headache, eight cases of limb paralysis, six cases of convulsions, and nine cases of coma, including three cases of immediate coma. Lumbar punctures were performed in six cases and all six cases had bloody CSF, which indicated that the onset of the CAA-related CLH was sudden and likely manifested with headache, paralysis, and unconsciousness. These manifestations may also include convulsions. The lumbar puncture examinations revealed that most cases had bloody CSF, which resulted from the hematoma breaking and entering the subarachnoid space or the ventricles. None of the patients ever exhibited systematic intelligence or were tested for cognitive function during their lifetime. In this study, eight of the 10 cases had CAA-related multiple CLH. The bleeding volume in nine cases was higher than 50 mL, and these cases died within several hours to days due to brain hernia. These results suggested that CAA-related CLH were usually multiple and fatal.
In addition to CLH, dementia and Alzheimer's disease (AD) are other diseases that are closely linked to CAA14). In a large study, one-third of the patients with pathologically confirmed CAA had a history of dementia11). Patients with CAA-related cerebral hemorrhage tend to have high incidence rates of AD and often carry the apolipoprotein E (APOE) ε4 allele, which is closely related to CAA. In addition, they overexpress the APOEε2 allele21). The ε2 allele is associated with cellulose necrosis and concentric splitting of the vascular wall (the double-barrel sign). Patients with CAA-related cerebral hemorrhage and the ε2/ε4 genotype have early onset and recurrent lobar hemorrhage26). It can be assumed that a complex gene-environment interaction exists in these patients with intracranial hemorrhages. In this study, eight of the 10 cases with CAA-related CLH (80%) had different degrees of NFTs, whereas five cases (50%) had SPs, indicating that CAA is closely related to SPs and NFTs. However, systematic evaluations of cognitive function were not performed when the patients were alive. Pathological studies have shown that, although AD and CAA-related cerebral hemorrhage often coexist, each disease can exist independently. Thus, AD and CAA-related cerebral hemorrhage are the outcomes of different Aβ pathological routes7).
The diagnosis of CAA
Rosand et al.33) applied gradient-echo magnetic resonance imaging (MRI) to detect 321 hemorrhage sites in 59 patients over 55 years of ages who were potentially suffering from CAA-related intracranial hemorrhage. They observed that the temporal and occipital lobes were the most common sites of bleeding, and bleeding easily recurred at the previous sites. These results were not consistent with previous MRI and autopsy results that showed that the occipital lobe was the characteristic site of CAA-related cerebral hemorrhage25). In the present study, CAA-related CLH was found most in the parietal, temporal, and occipital lobes and less in the frontal lobe. The distribution of CAA-related CLH indicated that certain characteristics of arteries, such as their branching patterns or susceptibility to arteriosclerosis, might affect their susceptibility to CAA-induced vascular bleeding disease14). The discrepancies between the CAA-related CLH distributions reported in in vivo MRI studies and at autopsy must be examined with extensive clinical and pathological studies34).
The exact pathogenesis of amyloid deposition-induced vascular rupture remains unclear. However, studies have found that, compared with nonbleeding CAA, patients with CAA-related cerebral hemorrhage often present with complications due to the CAA vascular lesions, which may be related to bleeding susceptibility30). These complications include the swelling and expansion of tiny aneurysms, fibrinoid necrosis, the double barrel-like appearance, and bleeding. In rare cases, vascular occlusion and vasculitis are observed. Fibrinoid necrosis and aneurysm formation may participate in the pathogenesis of vascular rupture in patients with CAA-related cerebral hemorrhage20). Maeda et al.19) used three-dimensional computer-aided image analysis and immunohistochemical methods and found that vascular segments with severe amyloidosis have a spider-like capillary hemangioma expansion. In these cases, the tunica media and tunica externa were almost completely replaced by amyloid. The elastic membrane and smooth muscle of the tunica media disappeared, which was thought to be the reason for microaneurysm formation8). In the severely expanded part of the hemangioma, the tunica media and tunica externa became thinner, whereas the tunica intima was transparently thickened. This may indicate a reactive change that is caused by amyloid-deposition-induced damage to the tunica media. The thickened tunica intima exhibits fibrinoid necrosis23). The immunohistochemistry results indicated the presence of fibrinogen or fibrin, and this was considered to be the pathogenesis of CAA-related cerebral hemorrhage9). In CAA, the cerebral cortex may also exhibit SPs, NFTs, and nerve cell loss. However, the exact mechanism by which amyloid deposition may promote vascular pathological changes remains unclear32).
In this study, two of the 10 cases had microaneurysms and four had double barrel-like changes. The term double barrel refers to intracellular concentric angiogenesis. In these double barrel changes, the vascular outer layer can be stained with Congo red, and the newly formed tunica externa may have amyloid infiltration. Loose connective tissue and mucosal substances can be observed in the gap of the double barrel. Five cases had multiple small arterial plexus changes, four had onion-like occlusive intimal changes, and seven had vascular wall-like cellulose necrosis. Cortical gliosis and neuronal degeneration were also observed. These CAA-associated microvasculopathy (CAA-AV) changes were more common in the cerebral cortex and meninges. In contrast, changes were rare in the basal ganglia and cerebellum, which may provide insight regarding the vasculopathy underlying CAA-related CLH. Although hypertensive patients occasionally exhibit these secondary vascular changes, the predilection sites of CAA-AV, with or without mild cerebral arteriosclerosis, and the simultaneous occurrence of CAA are distinct from hypertensive-secondary vascular changes. CAA lesions are mainly located in the meninges and brain cortex. The pathological features of CAA include amyloid deposition onto the vascular walls, whereas cerebral arteriosclerosis involves the hyaline degeneration of arterial walls and the deposition of cholesterol crystals.
The prognosis of CAA
The prognosis for most CAA patients is poor : 20% to 90% of patients die from the onset of bleeding or its complications, which include bleeding progression, cerebral edema-induced brain hernia, epilepsy, and infections, such as pneumonia. Most survivors have neurological deficits that are secondary to the lobar hemorrhage as well as an increased risk of recurrent bleeding, epilepsy, and dementia. Elderly patients and patients suffering from heavy bleeding or short-term recurrent bleeding have the worst prognosis.
In addition, CAA-related cerebral hemorrhages must be distinguished from other types of cerebral hemorrhage38). CAA-related hemorrhages typically occur in the cortical lobes, frequently break into the subarachnoid space between the brains and cover film, and often occur at night. Hypertensive hemorrhages usually occur deep in the brain, break into the ventricle or deep brain cavity, and normally occur in daytime40). The indications are typically indistinct in MRI scans of elderly patients with cognitive dysfunction, patients suffering from nontraumatic nonhypertensive cerebral hemorrhages in superficial areas, and, especially, patients with multiple CLH. In these cases, it would be beneficial to take the presence of CAA into account. Brain computed tomography (CT) or MRI may detect CLH, stroke, or spotted bleeding. Angiography is not effective for the diagnosis of CAA, but it can be useful for excluding aneurysms. Gradient-echo MRI can be applied to detect multiple asymptomatic lobar petechial hemorrhages, which are a typical characteristic of the disease, thereby enabling the in vivo diagnosis of suspected CAA16).
Because this study used 10-µm-thick sections and had a smaller sample size, there was a possibility of false negative results. There could be more neuropathological findings, including NFTs, SPs, and granular degeneration, in slices. However, changes that were related to blood vessel pathology, such as microaneurysms, double barrel-like changes, multiple small arterial plexus changes, onion-like endometrial changes, and blood vascular-type cellulose wall necrosis, were not likely to be missed.
This study had some limitations. A small number of cases were examined, and it was a retrospective study. Furthermore, there was the potential for a selection bias because all of the patients had severe clinical situations because they all died.
 XML Download
XML Download